




DEPARTMENT OF HEALTH AND SOCIAL SERVICES
Division of Public Health
PROPOSED
PUBLIC NOTICE
4465 Delaware Radiation Control Regulations
On July 1, 2017, the Department of Health and Social Services, Division of Public Health, Office of Radiation Control, plans to publish revised Regulations Governing Radiation Control - Part F and Part H and hold them out for public comment per Delaware law.
NOTICE OF PUBLIC HEARING
A public hearing will be held on Thursday, July 27, 2017, at 10:00 a.m. in the First Floor Conference Room, located in the Jesse Cooper Building, 417 Federal Street, Dover, Delaware.
The Authority on Radiation Protection (ARP), with the Office of Radiation Control, Health Systems Protection Section, Division of Public Health, Department of Health and Social Services, is proposing to repeal and replace two chapters of Delaware Radiation Control Regulations. The purpose of the amendments is to update the requirements so that they are in concert with current healthcare and industry standards, and to align them more closely with current state administrative code and federal requirements. The regulations will apply to any facility or person that receives, possesses, uses, transfers, sells, owns or acquires ionizing radiation sources, or provides radiation services to such radiation source facilities, or who administers machine-generated radiation to human patients in the healing arts.
Any person who wishes to make written suggestions, testimony, briefs or other written materials concerning the proposed regulation should submit such comments by Friday, August 11, 2017 to:
Jamie Mack, Executive Assistant
Office of the Director
Delaware Division of Public Health
Jesse Cooper Building
417 Federal St.
Dover, DE 19901
Email: jamie.mack@state.de.us
Fax: 302-739-3984
4465 Delaware Radiation Control Regulations
Part A General Provisions
Except as otherwise specifically provided, these regulations apply to all persons who receive, possess, use, transfer, own, or acquire any source of ionizing radiation. However, nothing in these regulations except for registration of radiation machine facilities/sources as specified in Regulation 4465 Part B shall apply to any person to the extent such person is subject to regulation by the Nuclear Regulatory Commission. See 4465 Parts C & G of these regulations which pertain to radioactive materials licensing and federal oversight.
As used in these regulations, these terms have the definitions set forth below. Additional definitions used only in a certain Part will be found in that Part.
"A1" means the maximum activity of special form radioactive material permitted in a Type A package. "A2" means the maximum activity of radioactive material, other than special form radioactive material, permitted in a Type A package. These values are either listed in Appendix A of Part T of these regulations, Table I, or may be derived in accordance with the procedure prescribed in Appendix A of Part T of these regulations.
"Absorbed dose" means the energy imparted by ionizing radiation per unit mass of irradiated material. The units of absorbed dose are the gray (Gy) and the rad.
"Accelerator" means any machine capable of accelerating electrons, protons, deuterons, or other charged particles in a vacuum and of discharging the resultant particulate or other radiation into a medium at energies usually in excess of 1 MeV. For purposes of this definition, "particle accelerator" is an equivalent term.
"Accelerator-produced material" means any material made radioactive by a particle accelerator.
"Activity" means the rate of disintegration or transformation or decay of radioactive material. The units of activity are the becquerel (Bq) and the curie (Ci).
“Address of use” means the building or buildings that are identified on the permit (license) and where radioactive materials may be produced, prepared, received, used, or stored.
"Adult" means an individual 18 or more years of age.
“Agency” means the Division of Public Health, Delaware Department of Health and Social Services.
"Agreement State" means any State with which the Nuclear Regulatory Commission or the Atomic Energy Commission has entered into an effective agreement under subsection 274b. of the Atomic Energy Act of 1954, as amended (73 Stat. 689).
"Airborne radioactive material" means any radioactive material dispersed in the air in the form of dusts, fumes, particulates, mists, vapors, or gases.
"Airborne radioactivity area" means a room, enclosure, or area in which airborne radioactive materials exist in concentrations:
(1) In excess of the derived air concentrations (DAC's) specified in Appendix B, Table I of Part D of these regulations; or
(2) To such a degree that an individual present in the area without respiratory protective equipment could exceed, during the hours an individual is present in a week, an intake of 0.6 percent of the annual limit on intake (ALI) or 12 DAC-hours.
"Airline respirator" (see"Supplied-air respirator (SAR)").
"Air-purifying respirator" means a respirator with an air-purifying filter, cartridge, or canister that removes specific air contaminants by passing ambient air through the air-purifying element.
“As low as is reasonably achievable" (ALARA) means making every reasonable effort to maintain exposures to radiation as far below the dose limits in these regulations as is practical, consistent with the purpose for which the licensed or registered activity is undertaken, taking into account the state of technology, the economics of improvements in relation to state of technology, the economics of improvements in relation to benefits to the public health and safety, and other societal and socioeconomic considerations, and in relation to utilization of nuclear energy and licensed or registered sources of radiation in the public interest.
"Assigned Protection Factor (APF)" means the expected workplace level of respiratory protection that would be provided by a properly functioning respirator or a class of respirators to properly trained and fitted users. Operationally, the inhaled concentration can be estimated by dividing the ambient airborne concentration by the APF.
"Atmosphere-supplying respirator" means a respirator that supplies the respirator user with breathing air from a source independent of the ambient atmosphere, and includes supplied-air respirators (SAR’s) and self-contained breathing apparatus (SCBA) units.
“Authorized user” means a practitioner of the healing arts who is identified as an authorized user on an Agency, Agreement State, Licensing State or the Nuclear Regulatory Commission license that authorizes the medical use of radioactive material.
"Background radiation" means radiation from cosmic sources, naturally occurring radioactive material, (which has not been technologically enhanced) including radon, except as a decay product of source or special nuclear material, and including global fallout as it exists in the environment from the testing of nuclear explosive devices, or from past nuclear accidents such as Chernobyl that contribute to background radiation and are not under the control of the licensee or registrant. "Background radiation" does not include sources of radiation from radioactive materials regulated by the Agency.
"Becquerel" (Bq) means the Standard Internationale (SI) unit of activity. One becquerel is equal to 1 disintegration or transformation per second (dps or tps).
"Bioassay" means the determination of kinds, quantities or concentrations, and, in some cases, the locations of radioactive material in the human body, whether by direct measurement, in vivo counting, or by analysis and evaluation of materials excreted or removed from the human body. For purposes of these regulations, "radiobioassay" is an equivalent term.
"Brachytherapy" means a method of radiation therapy in which radiation sources are utilized to deliver a radiation dose at a distance of up to a few centimeters, by surface, intracavitary, intraluminal, or interstitial application.
"Byproduct material" means:
(1) Any radioactive material (except special nuclear material) yielded in, or made radioactive by, exposure to the radiation incident to the process of producing or using special nuclear material;
(2) The tailings or wastes produced by the extraction or concentration of uranium or thorium from ore processed primarily for its source material content, including discrete surface wastes resulting from uranium solution extraction processes. Underground ore bodies depleted by these solution extraction operations do not constitute "byproduct material" within this definition;
(3) (i) Any discrete source of radium-226 that is produced, extracted, or converted after extraction, before, on, or after August 8, 2005, for use for a commercial, medical, or research activity; or
(ii) Any material that—
(A) Has been made radioactive by use of a particle accelerator; and
(B) Is produced, extracted, or converted after extraction, before, on, or after August 8, 2005, for use for a commercial, medical, or research activity; and
(4) Any discrete source of naturally occurring radioactive material, other than source material, that—
(i) The Commission, in consultation with the Administrator of the Environmental Protection Agency, the Secretary of Energy, the Secretary of Homeland Security, and the head of any other appropriate Federal agency, determines would pose a threat similar to the threat posed by a discrete source of radium-226 to the public health and safety or the common defense and security; and
(ii) Before, on, or after August 8, 2005, is extracted or converted after extraction for use in a commercial, medical, or research activity.
"Calendar quarter" means not less than 12 consecutive weeks nor more than 14 consecutive weeks. The first calendar quarter of each year shall begin in January and subsequent calendar quarters shall be so arranged such that no day is included in more than one calendar quarter and no day in any one year is omitted from inclusion within a calendar quarter. The method observed by the licensee or registrant for determining calendar quarters shall only be changed at the beginning of a year.
"Calibration" means the determination of (1) the response or reading of an instrument relative to a series of known radiation values over the range of the instrument, or (2) the strength of a source of radiation relative to a standard.
"CFR" means Code of Federal Regulations.
“Chiropractic” means a drugless system of health care based on the principle that interference with the transmission of nerve impulses may cause disease, per Title 24 Delaware Code, Chapter 7, Board of Chiropractic, as amended.
"Collective dose" means the sum of the individual doses received in a given period of time by a specified population from exposure to a specified source of radiation.
"Committed dose equivalent" (HT.50) means the dose equivalent to organs or tissues of reference (T) that will be received from an intake of radioactive material by an individual during the 50-year period following the intake.
"Committed effective dose equivalent" (HE.50) is the sum of the products of the weighting factors (wT) applicable to each of the body organs or tissues that are irradiated and the committed dose equivalent to each of these organs or tissues (HE,50 = Σ wT HT,50).
“Controlled area” means an area, outside of a restricted but inside the site boundary, access to which can be limited by the licensee or registrant, for any reason.
“Critical group” means the group of individuals reasonably expected to receive the greatest exposure to residual radioactivity for any applicable set of circumstances.
"Curie" means the traditional unit of quantity of activity. One curie (Ci) is that quantity of radioactive material, which decays at the rate of 3.7E+10 disintegrations or transformations per second (dps or tps).
"Deep dose equivalent" (Hd), which applies to external whole body exposure, means the dose equivalent at a tissue depth of 1 centimeter (1000 mg/cm2).
"Demand respirator" means an atmosphere-supplying respirator that admits breathing air to the face piece only when a negative pressure is created inside the facepiece by inhalation
“Dentist” shall mean a person who is qualified to practice dentistry as prescribed in Title 24 Delaware Code, Chapter 11, Dentistry and Dental Hygiene, as amended.
"Department of Energy" means the Department of Energy established by Public Law 95-91, August 4, 1977, 91 Stat. 565, 42 U.S.C. Section 7101 as amended et seq., to the extent that the Department exercises functions formerly vested in the Atomic Energy Commission, its Chairman, members, officers and components and transferred to the Energy Research and Development Administration and to the Administrator thereof pursuant to sections 104(b), (c) and (d) of the Energy Reorganization Act of 1974 (Public Law 93-438, October 11, 1974, 88 Stat. 1233 at 1237, 42 U.S.C. 5814, effective January 19, 1975) and re-transferred to the Secretary of Energy pursuant to section 301(a) of the Department of Energy Organization Act (Public Law 95-91, August 4, 1977, 91 Stat. 565 at 577-578, 42 U.S.C. 7151, effective October 1, 1977 as amended.)
"Depleted uranium" means the source material uranium in which the isotope uranium-235 is less than 0.711 weight percent of the total uranium present. Depleted uranium does not include special nuclear material.
“Discrete Source” means a radionuclide that has been processed so that its concentration within a material has been purposely increased for use for commercial, medical, or research activities.
"Disposable respirator" means a respirator for which maintenance is not intended and that is designed to be discarded after excessive breathing resistance, sorbent exhaustion, physical damage, or end-of-service-life renders it unsuitable for use. Examples of this type of respirator are a disposable half-mask respirator or a disposable escape-only self-contained breathing apparatus (SCBA).
"Distinguishable from background" means that the detectable concentration of a radionuclide is statistically different from the background concentration of that radionuclide in the vicinity of the site or, in the case of structures, in similar materials using adequate measurement technology, survey, and statistical techniques.
"Dose" is a generic term that means absorbed dose, dose equivalent, effective dose equivalent, committed dose equivalent, committed effective dose equivalent, total organ dose equivalent, or total effective dose equivalent. For purposes of these regulations, "radiation dose" is an equivalent term.
"Dose equivalent (HT)" means the product of the absorbed dose in tissue, quality factor, and all other necessary modifying factors at the location of interest. The units of dose equivalent are the sievert (Sv) and rem.
"Dose limits" means the permissible upper bounds of radiation doses established in accordance with these regulations. For purposes of these regulations, "limits" is an equivalent term.
"Effective dose equivalent (HE)" means the sum of the products of the dose equivalent to the organ or tissue (HT) and the weighting factor (wT) applicable to each of the body organs or tissues that are irradiated (HE = Σ wTHT).
"Embryo/fetus" means the developing human organism from conception until the time of birth.
"Exposure" generally means being exposed to ionizing radiation or to radioactive material;
"Exposure Units" specifically as used in these regulations, the SI unit of exposure is coulomb per kilogram (C/kg), see Section A.9.1 of this Part for Units of Exposure and Dose.
"Exposure rate" means the exposure per unit of time, such as roentgen per minute or milliroentgen per hour.
"External dose" means that portion of the dose equivalent received from any source of radiation outside the body.
"Extremity" means hand, elbow, and arm below the elbow, foot, knee, and leg below the knee.
“Facility” means the location, building vehicle, or complex under one administrative control, at which one or more radiation sources are installed, located and/or used.
"Filtering facepiece (dust mask)" means a negative pressure particulate respirator with a filter as an integral part of the facepiece or with the entire facepiece composed of the filtering medium, not equipped with elastomeric sealing surfaces and adjustable straps.
"Fit factor" means a quantitative estimate of the fit of a particular respirator to a specific individual, and typically estimates the ratio of the concentration of a substance in ambient air to its concentration inside the respirator when worn.
"Fit Test" means the use of a protocol to qualitatively evaluate the fit of a respirator on an individual.
"Former Atomic Energy Commission or Nuclear Regulatory Commission licensed facilities" means nuclear reactors, nuclear fuel reprocessing plants, uranium enrichment plants, or critical mass experimental facilities where Atomic Energy Commission or Nuclear Regulatory Commission licenses have been terminated.
"Generally applicable environmental radiation standards" means standards issued by the Environmental Protection Agency under the authority of the Atomic Energy Act of 1954, as amended, that impose limits on radiation exposures or levels, or concentrations or quantities of radioactive material, in the general environment outside the boundaries of locations under the control of persons possessing or using radioactive material.
"Gray" (Gy) means the Standard Internationale (SI) unit of absorbed dose. One gray is equal to an absorbed dose of 1 joule per kilogram (100 rad).
"Hazardous waste" means those wastes designated as hazardous by the Environmental Protection Agency regulations in 40 CFR Part 261, as amended.
“Healing arts” includes but is not limited to the practice of medicine, surgery, dentistry, registered pharmacy, podiatry, osteopathy, chiropractic, or veterinary medicine or nursing.
“Healing arts screening” means the testing of human beings using x-ray machines for the detection or evaluation of health indications when such tests are not specifically and individually ordered by a licensed practitioner of the healing arts legally authorized to prescribe such x-ray tests for the purpose of diagnosis or treatment.
"Helmet" means a rigid respiratory inlet covering that also provides head protection against impact and penetration.
"High radiation area" means an area, accessible to individuals, in which radiation levels from radiation sources external to the body could result in an individual receiving a dose equivalent in excess of 1 mSv (0.1 rem) in 1 hour at 30 centimeters from any source of radiation or 30 centimeters from any surface that the radiation penetrates.
"Hood" means a respiratory inlet covering that completely covers the head and neck and may also cover portions of the shoulders and torso.
"Human use" means the internal or external administration of radiation or radioactive material to human beings.
"Individual" means any human being.
"Individual monitoring" means the assessment of:
(1) Dose equivalent (a) by the use of individual monitoring devices or (b) by the use of survey data; or
(2) Committed effective dose equivalent (a) by bioassay or (b) by determination of the time-weighted air concentrations to which an individual has been exposed, that is, DAC-hours. [See the definition of DAC-hours in 4465 Part D of these regulations.]
(3) Dose equivalent by the use of survey data.
"Individual monitoring devices" means devices designed to be worn by a single individual for the assessment of dose equivalent. For purposes of these regulations, "personnel dosimeter" and "dosimeter" are equivalent terms. Examples of individual monitoring devices are film badges, thermoluminescence dosimeters (TLDs), pocket ionization chambers, optically stimulated luminescence (OSL) dosimeters and personal (lapel) air sampling devices.
"Inspection" means an official examination or observation including, but not limited to, tests, surveys, and monitoring to determine compliance with rules, regulations, orders, requirements, and conditions of the Agency.
"Instrument traceability" (for ionizing radiation measurements) means the ability to show that an instrument has been calibrated at specified time intervals using a national standard or a transfer standard. If a transfer standard is used, the calibration must be at a laboratory accredited by a program, which requires continuing participation in measurement quality assurance with the National Institute of Standards and Technology, or other equivalent national or international program.
"Interlock" means a device arranged or connected such that the occurrence of an event or condition is required before a second event or condition can occur or continue to occur.
"Internal dose" means that portion of the dose equivalent received from radioactive material taken into the body.
"JRCECT" means Joint Review Committee on Education in Cardiovascular Technology
“JRCNMT” means Joint Review Committee on Nuclear Medicine Technology
"JRCERT" means Joint Review Committee on Education in Radiologic Technology
"Lens dose equivalent (LDE)" means the external exposure to the lens of the eye as the dose equivalent at a tissue depth of 0.3 centimeter (300 mg/cm2).
"License" means a license issued by the US Nuclear Regulatory Commission, Agreement State, or the Agency, in accordance with applicable federal or state regulations, as amended.
“Licensed Practitioner” means a physician licensed to practice medicine, dentistry, podiatry, chiropractic, osteopathy, or veterinary medicine in this state.
"Licensed Practitioner" means an individual licensed to practice medicine, dentistry, podiatry, chiropractic, osteopathy, advanced practice nursing, or veterinary medicine in this state.
"Licensed [or registered] material" means radioactive material received, possessed, used, transferred or disposed of under a general or specific license [or registration] issued by the Agency.
"Licensee" means the holder of a license.
"Limits" [See "Dose limits"].
"Loose-fitting facepiece" means a respiratory inlet covering that is designed to form a partial seal with the face.
"Lost or missing source of radiation" means licensed [or registered] source of radiation whose location is unknown. This definition includes, but is not limited to, radioactive material that has been shipped but has not reached its planned destination and whose location cannot be readily traced in the transportation system.
"Major processor" means a user processing, handling, or manufacturing radioactive material exceeding Type A quantities as unsealed sources or material, or exceeding 4 times Type B quantities as sealed sources, but does not include nuclear medicine programs, universities, industrial radiographers, or small industrial programs. Type A and B quantities are defined in T.2 of these regulations.
"Member of the public" means any individual except when that individual is receiving an occupational dose.
"Minor" means an individual less than 18 years of age.
“Misadministration” means an event that meets the criteria in 4465 Part X, Therapeutic Radiation Machines, Section 5.2 of these regulations.
"Monitoring" means the measurement of radiation, radioactive material concentrations, surface area activities or quantities of radioactive material and the use of the results of these measurements to evaluate potential exposures and doses. For purposes of these regulations, "radiation monitoring" and "radiation protection monitoring" are equivalent terms.
"Natural radioactivity" means radioactivity of naturally occurring nuclides.
"Negative pressure respirator (tight fitting)" means a respirator in which the air pressure inside the facepiece is negative during inhalation with respect to the ambient air pressure outside the respirator.
"NORM" means any naturally occurring radioactive material. It does not include byproduct, source, or special nuclear material.
"NRC" means the US Nuclear Regulatory Commission or its duly authorized representatives.
“Notice of Violation” means a written statement of one or more alleged infringements of a legally binding requirement. The notice normally requires the licensee, registrant or other permit holder to provide a written statement describing the following:
(1) Corrective steps taken by the licensee, registrant or other permit holder and the results achieved;
(2) Corrective steps to be taken to prevent recurrence; and
(3) The projected date for achieving full compliance. The Authority may require responses to notices of violation to be under oath.
"Occupational dose" means the dose received by an individual in the course of employment in which the individual's assigned duties for the licensee or registrant involve exposure to sources of radiation, whether or not the sources of radiation are in the possession of the licensee, registrant, or other person. Occupational dose does not include doses received from background radiation, or from any medical administration the individual has received, from exposure to individuals administered radioactive material and released in accordance with U.S. Nuclear Regulatory Commission Regulations, from voluntary participation in medical research programs, or as a member of the public.
“Office of Engineering” means the office in the Delaware Division of Public Health that reviews radiation shielding plans and/or design plans and issues an Approval to Construct letter for new radiation source facilities or rooms.
“Office of Radiation Control” means the office in the Delaware Division of Public Health which carries out the Delaware Radiation Control Regulations, issues radiation source facility registration permits, and performs on-site inspections of new and existing radiation machine facilities to determine compliance.
“Owner/Leasee” means the person/individual who owns/leases the radiation source. An out-of-state owner shall authorize a manager to sign the application form.
"Package" means the packaging together with its radioactive contents as presented for transport.
"Particle accelerator" [See "Accelerator"].
"Person" means any individual, corporation, partnership, firm, association, trust, estate, public or private institution, group, agency, political subdivision of this State, any other State or political subdivision or agency thereof, and any legal successor, representative, agent, or agency of the foregoing, [but shall not include federal government agencies].
"Personnel monitoring equipment" [See "Individual monitoring devices"].
"Physician" means an allopathic doctor of medicine and surgery or a doctor of osteopathic medicine and surgery who is registered and certified to practice medicine pursuant to Title 24 Delaware Code, Chapter 17, Medical Practice Act, as amended.
“Podiatrist” means a person who is qualified to practice podiatry and is licensed under Title 24 Delaware Code, Chapter 5, Podiatry, as amended.
"Positive pressure respirator" means a respirator in which the pressure inside the respiratory inlet covering exceeds the ambient air pressure outside the respirator.
"Powered air-purifying respirator (PAPR)" means an air-purifying respirator that uses a blower to force the ambient air through air-purifying elements to the inlet covering.
"Pressure demand respirator" means a positive pressure atmosphere-supplying respirator that admits breathing air to the facepiece when the positive pressure is reduced inside the facepiece by inhalation.
“Principal Supervisor” means the State-Licensed Practitioner responsible for initiating use of x-ray equipment or other device generating ionizing radiation in the healing arts.
"Protective apron" means an apron made of radiation-attenuating materials used to reduce exposure to radiation.
"Public dose" means the dose received by a member of the public from exposure to sources of radiation released by the licensee or registrant, or to any other source of radiation under the control of the licensee or registrant. Public dose does not include occupational dose, or doses received from background radiation, from any medical administration the individual has received, from exposure to individuals administered radioactive material and released in accordance with U.S. Nuclear Regulatory Commission Regulations, or from voluntary participation in medical research programs.
“Qualified expert” means an individual who has satisfactorily fulfilled the training and experience requirements consistent with achieving a level of competency sufficient to function effectively in the position for which registration is sought. Such individuals must demonstrate to the satisfaction of the Agency their qualifications, for example, individuals certified in the appropriate field by the American Board of Radiology, or the American Board of Health Physics, or the American Board of Medical Physics, or those having equivalent qualifications. With reference to the calibration of radiation therapy equipment, an individual, in addition to the above qualifications, must be qualified in accordance with 4465 Part F and 4465 Part X of these regulations, as amended.
“Qualified Medical Physicist” means an individual qualified in accordance with Regulation 4465, Part X, Therapeutic Radiation Machines, Section 3.4, as amended. means an individual who meets each of the following credentials:
1. Has earned a master's and/or doctoral degree in physics, medical physics, biophysics, radiological physics, medical health physics, or equivalent disciplines from an accredited college or university; and
2. Has been granted certification in the specific subfield(s) of medical physics with its associated medical health physics aspects by an appropriate national certifying body and abides by the certifying body's requirements for continuing education; and/or
3. Is credentialed in accordance with Part X, subsection 3.4, as amended.
"Qualitative fit test (QLFT)" means a pass/fail fit test to assess the adequacy of respirator fit that relies on the individual’s response to the test agent.
"Quality factor" (Q) means the modifying factor, listed in Tables I and II of A.13, that is used to derive dose equivalent from absorbed dose.
"Quantitative fit test (QNFT)" means an assessment of the adequacy of respirator fit by numerically measuring the amount of leakage into the respirator.
"Rad" means the traditional unit of absorbed dose. One rad is equal to an absorbed dose of 100 erg per gram or 0.01 joule per kilogram (0.01 gray).
"Radiation" means alpha particles, beta particles, gamma rays, x rays, neutrons, high-speed electrons, high-speed protons, and other particles capable of producing ions. For purposes of these regulations, ionizing radiation is an equivalent term. Radiation, as used in these regulations, does not include non-ionizing radiation, such as radiowaves or microwaves, visible, infrared, or ultraviolet light.
"Radiation area" means any area, accessible to individuals, in which radiation levels could result in an individual receiving a dose equivalent in excess of 0.05 mSv (0.005 rem) in 1 hour at 30 centimeters from the source of radiation or from any surface that the radiation penetrates.
"Radiation dose" [See "Dose"].
"Radiation machine" means any device capable of producing ionizing radiation except those devices with radioactive material as the only source of radiation.
“Radiation Safety Officer” or RSO for a radiation machine facility means an individual assigned to perform RSO duties who has training and experience in the safe and effective use of radiation machines, their potential radiation hazards, and emergency precautions applicable to the type of activity or facility to which the RSO is assigned.
"Radiation Technician" means any individual who has not graduated from an approved program in radiation technology, but has passed an Authority approved examination.
"Radiation Technologist" means any individual who has successfully completed a JRCERT/JRCCVT approved program in radiation technology and has passed a national certification examination in his/her field of specialization.
"Radiation Technology" means the use of a radioactive substance or equipment emitting ionizing radiation on humans for diagnostic or therapeutic purposes.
"Radioactive material" means any solid, liquid or gas which emits radiation spontaneously.
"Radioactivity" means the transformation of unstable atomic nuclei by the emission of radiation.
"Radiobioassay" [See "Bioassay"].
"Registrant" means any person who is registered with the Agency and is legally obligated to register with the Agency pursuant to these regulations and the Act.
"Registration" means registration with the Agency in accordance with the regulations adopted by the Agency.
"Regulations of the Department of Transportation" means the regulations in 49 CFR Parts 100-189, as amended.
"Rem" means the traditional unit of any of the quantities expressed as dose equivalent. The dose equivalent in rem is equal to the absorbed dose in rad multiplied by the quality factor. (1 rem = 0.01 Sv)
"Research and development" means (1) theoretical analysis, exploration, or experimentation; or (2) the extension of investigative findings and theories of a scientific or technical nature into practical application for experimental and demonstration purposes, including the experimental production and testing of models, devices, equipment, materials, and processes. Research and development does not include the internal or external administration of radiation or radioactive material to human beings in the healing arts.
"Residual radioactivity" means radioactivity in structures, materials, soils, groundwater, and other media at a site resulting from activities under the licensee’s control. This includes radioactivity from all licensed and unlicensed sources used by the licensee, but excludes background radiation. It also includes radioactive materials remaining at the site as a result of routine or accidental releases of radioactive materials at the site and previous burials at the site, even if those burials were made in accordance with the provisions of Part D of these regulations.
"Restricted area" means an area, access to which is limited by the licensee or registrant for the purpose of protecting individuals against undue risks from exposure to sources of radiation. Restricted area does not include areas used as residential quarters, but separate rooms in a residential building may be set apart as a restricted area.
"Roentgen" means the traditional unit of exposure. One roentgen (R) equals 2.58E-4 coulombs per kilogram of air (see "Exposure" and Part A.9.1 of this part.)
“State Radiation Control Act” or “the Act” means Title 16 Delaware Code, Chapter 74, Radiation Control, as amended.
"Sealed source" means any encapsulated radioactive material, which has been constructed in such a manner as to prevent the escape of any radioactive material.
"Sealed Source and Device Registry (SSD)" means the national registry that contains the registration certificates, maintained by the Nuclear Regulatory Commission (NRC), that summarize the radiation safety information for sealed sources and devices, and describe the licensing and use conditions approved for the product.
"Self-contained breathing apparatus (SCBA)" means an atmosphere-supplying respirator for which the breathing air source is designed to be carried by the user.
"Shallow dose equivalent" (Hs), which applies to the external exposure of the skin or an extremity, means the dose equivalent at a tissue depth of 0.007 centimeter (7 mg/cm2) averaged over the contiguous 10 square centimeters of skin receiving the highest exposure.
"SI" means the abbreviation for Standard Internationale, the International Metric System of Measurement.
"Sievert" means the Standard Internationale (SI) unit of any of the quantities expressed as dose equivalent. The dose equivalent in sievert is equal to the absorbed dose in gray multiplied by the quality factor. (1 Sv = 100 rem)
"Source material" means:
(1) Uranium or thorium, or any combination thereof, in any physical or chemical form; or
(2) Ores that contain by weight one-twentieth of 1 percent (0.05 percent) or more of uranium, thorium or any combination of uranium and thorium. Source material does not include special nuclear material.
"Source material milling" means any activity that results in the production of byproduct material as defined by definition (2) of byproduct material, of this part.
"Source of radiation" means any radioactive material or any device or equipment emitting, or capable of producing, radiation.
"Source traceability" means the ability to show that a radioactive source has been calibrated either by the national standards laboratory of the National Institute of Standards and Technology, or by a laboratory which participates in a continuing measurement quality assurance program with National Institute of Standards and Technology or other equivalent national or international program.
"Special form radioactive material" means radioactive material that satisfies the following conditions:
(1) It is either a single solid piece or is contained in a sealed capsule that can be opened only by destroying the capsule;
(2) The piece or capsule has at least one dimension not less than 5 millimeters (0.2 inch); and
(3) It satisfies the test requirements specified by the Nuclear Regulatory Commission. A special form encapsulation designed in accordance with the Nuclear Regulatory Commission requirements in effect on June 30, 1983, and constructed prior to July 1, 1985, may continue to be used. A special form encapsulation either designed or constructed after June 30, 1985, must meet requirements of this definition applicable at the time of its design or construction.
"Special nuclear material" means:
(1) Plutonium, uranium-233, uranium enriched in the isotope 233 or in the isotope 235, and any other material that the Nuclear Regulatory Commission, pursuant to the provisions of section 51 of the Atomic Energy Act of 1954, as amended, determines to be special nuclear material, but does not include source material; or
(2) Any material artificially enriched by any of the foregoing but does not include source material.
"Special nuclear material in quantities not sufficient to form a critical mass" means uranium enriched in the isotope U-235 in quantities not exceeding 350 grams of contained U-235; uranium-233 in quantities not exceeding 200 grams; plutonium in quantities not exceeding 200 grams; or any combination of them in accordance with the following formula: For each kind of special nuclear material, determine the ratio between the quantity of that special nuclear material and the quantity specified above for the same kind of special nuclear material. The sum of such ratios for all of the kinds of special nuclear material in combination shall not exceed 1. For example, the following quantities in combination would not exceed the limitation and are within the formula: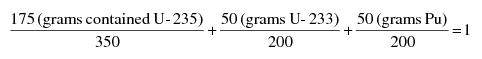
“Standard Internationale (SI)” means the international metric systems of measurement.
"Supplied-air respirator (SAR)" means an atmosphere-supplying respirator for which the source of breathing air is not designed to be carried by the user.
"Survey" means an evaluation of the radiological conditions and potential hazards incident to the production, use, transfer, release, disposal, or presence of sources of radiation. When appropriate, such evaluation includes, but is not limited to, tests, physical examinations, and measurements of levels of radiation or concentrations of radioactive material present.
"Test" means the process of verifying compliance with an applicable regulation.
"These regulations" means all parts of The Delaware Radiation Control Regulations 4465, as amended.
"Tight-fitting facepiece" means a respiratory inlet covering that forms a complete seal with the face.
"Total effective dose equivalent" (TEDE) means the sum of the deep dose equivalent for external exposures and the committed effective dose equivalent for internal exposures.
"Total organ dose equivalent" (TODE) means the sum of the deep dose equivalent and the committed dose equivalent to the organ receiving the highest dose as described in D.1107a.vi. of these regulations.
"Traceable to a National Standard" [See "Instrument traceability" or "Source traceability"].
"Unrefined and unprocessed ore" means ore in its natural form prior to any processing such as grinding, roasting, beneficiating, or refining.
"Unrestricted area" means an area, access to which is neither limited nor controlled by the licensee or registrant. For purposes of these regulations, "uncontrolled area" is an equivalent term.
"User seal check (fit check)" means an action conducted by the respirator user to determine if the respirator is properly seated to the face. Examples include negative pressure check, positive pressure check, irritant smoke check, or isoamyl acetate check.
"Very high radiation area" means an area, accessible to individuals, in which radiation levels from radiation sources external to the body could result in an individual receiving an absorbed dose in excess of 5 Gy (500 rad) in 1 hour at 1 meter from a source of radiation or 1 meter from any surface that the radiation penetrates.12/
“Veterinarian” shall mean a person who has received a degree in veterinary medicine from a school of veterinary medicine, per Title 24 Delaware Code, Chapter 33, Veterinarians, as amended.
"Waste" means those low-level radioactive wastes that are acceptable for disposal in a land disposal facility. For the purposes of this definition, low-level waste has the same meaning as in the Low-Level Radioactive Waste Policy Act, P.L. 96-573, as amended by P.L. 99-240, effective January 15, 1986; that is, radioactive waste (a) not classified as high-level radioactive waste, spent nuclear fuel, or byproduct material as defined in Section 11e.(2) of the Atomic Energy Act, as amended (uranium or thorium tailings and waste) and (b) classified as low-level radioactive waste consistent with existing law and in accordance with (a) by the Nuclear Regulatory Commission.
"Waste handling licensees" mean persons licensed to receive and store radioactive wastes prior to disposal and/or persons licensed to dispose of radioactive waste.
"Week" means 7 consecutive days starting on Sunday.
"Whole body" means, for purposes of external exposure, head, trunk including male gonads, arms above the elbow, or legs above the knee.
"Worker" means an individual engaged in activities under a license or registration issued by the Agency and controlled by a licensee or registrant, but does not include the licensee or registrant.
"Working level" (WL) means any combination of short-lived radon daughters in 1 liter of air that will result in the ultimate emission of 1.3E+5 MeV of potential alpha particle energy. The short-lived radon daughters of radon-222 are polonium-218, lead-214, bismuth-214, and polonium-214; and those of radon-220 are polonium-216, lead-212, bismuth-212, and polonium-212.
"Working level month" (WLM) means an exposure to 1 working level for 170 hours. 2,000 working hours per year divided by 12 months per year is approximately equal to 170 hours per month.
"Year" means the period of time beginning in January used to determine compliance with the provisions of these regulations. The licensee or registrant may change the starting date of the year used to determine compliance by the licensee or registrant provided that the change is made at the beginning of the year. If a licensee or registrant changes in a year, the licensee or registrant shall assure that no day is omitted or duplicated in consecutive years.
3.1 Exemptions. An exemption may be granted by the Agency if, based on documented and publicly available information, the Agency has verified that the proposed exempted practice or equipment does not pose any danger to the applicant, his employees or any others coming into contact with the exempted practice or equipment. An exemption request that deviates from accepted standards as specified in the regulations, such that the safe use of said practice or equipment cannot be supported by extraneous documented and publicly available information must be referred to the Authority on Radiation Protection for consideration.
3.1.1 General Provision. The Agency as the Agent for the Authority on Radiation Protection may, upon application or upon its own initiative, grant such exemptions or exceptions from the requirements of the regulations as it determines are authorized by law and will not result in undue hazard to public health and safety or property.
3.1.2 Department of Energy Contractors and Nuclear Regulatory Commission Contractors. Any Department of Energy contractor or subcontractor and any Nuclear Regulatory Commission contractor or subcontractor of the following categories operating within this State is exempt from the regulations to the extent that such contractor or subcontractor under his contract receives, possesses, uses, transfers, or acquires sources of radiation:
3.1.2.1 Prime contractors performing work for the Department of Energy at U.S. Government-owned or -controlled sites, including the transportation of sources of radiation to or from such sites and the performance of contract services during temporary interruptions of such transportation;
3.1.2.2 Prime contractors of the Department of Energy performing research in, or development, manufacture, storage, testing, or transportation of, atomic weapons or components thereof;
3.1.2.3 Prime contractors of the Department of Energy using or operating nuclear reactors or other nuclear devices in a United States Government-owned vehicle or vessel; and
3.1.2.4 Any other prime contractor or subcontractor of the Department of Energy or of the Nuclear Regulatory Commission when the State and the Nuclear Regulatory Commission jointly determine:
3.1.2.4.1 That the exemption of the prime contractor or subcontractor is authorized by law; and
3.1.2.4.2 That, under the terms of the contract or subcontract, there is adequate assurance that the work thereunder can be accomplished without undue risk to the public health and safety.
4.1 Records. Each licensee and registrant shall maintain records showing the receipt, transfer, and disposal of all sources of radiation. Additional record requirements are specified elsewhere in the regulations.
4.2 Inspections
4.2.1 Each licensee and registrant shall afford the Agency at all reasonable times opportunity to inspect sources of radiation and the premises and facilities wherein such sources of radiation are used or stored.
4.2.2 Each licensee and registrant shall make available to the Agency for inspection, upon reasonable notice, records maintained pursuant to the regulations.
4.3 Tests. Each licensee and registrant shall perform upon instructions from the Agency, or shall permit the Agency to perform, such reasonable tests as the Agency deems appropriate or necessary including, but not limited to, tests of:
4.3.1 Sources of radiation;
4.3.2 Facilities wherein sources of radiation are used or stored;
4.3.3 Radiation detection and monitoring instruments; and
4.3.4 Other equipment and devices used in connection with utilization or storage of licensed or registered sources of radiation.
The Authority through the Agency may, by rule, regulation, or order, impose upon any licensee or registrant such requirements in addition to those established in the regulations as it deems appropriate or necessary to minimize danger to public health and safety or property.
6.1 Violations. An injunction or other court order may be obtained prohibiting any violation of any provision of the State Radiation Control Act, as amended or any regulation or order issued thereunder. The Authority may request the Attorney General to make application to the Court of Chancery for an order enjoining any acts or practices which constitute or will constitute a violation of any provision of this chapter or any rule, regulation or order issued thereunder.
6.2 Impounding. Sources of radiation shall be subject to impoundment pursuant to Title 16 Delaware Code, Section 7415 of the State Radiation Control Act, as amended.
6.3 Prohibited Uses
6.3.1 A hand-held fluoroscopic screen shall not be used with x-ray equipment unless it has been listed in the Registry of Sealed Source and Devices or accepted for certification by the Food and Drug Administration, Center for Devices and Radiological Health.
6.3.2 A shoe-fitting fluoroscopic device shall not be used.
6.3.3 A closed end PID (conical position indicating device) shall not be used.
6.3.4 A source of radiation shall not be abandoned.
6.4 Penalties. In addition to any other remedies available to the Authority – the Authority may assess an administrative penalty in an amount not to exceed $500 for a first offense, an amount not to exceed $750 for a subsequent offense. Each violation of this chapter or rules, regulations or orders shall be considered a separate offense.
Except as specifically authorized by the Agency in writing, no interpretation of the regulations by an officer or employee of the Agency other than a written interpretation by the Authority on Radiation Protection will be recognized to be binding upon the Agency.
All communications and reports concerning the regulations, and applications filed thereunder, should be addressed to the Agency at its Office of Radiation Control, Division of Public Health, 417 Federal Street, Dover, DE 19901.
9.1 As used in these regulations, the unit of exposure is the coulomb per kilogram (C/kg) of air. One roentgen is equal to 2.58E-4 coulomb per kilogram of air.
9.2 As used in these regulations, the units of dose are:
9.2.1 Gray (Gy) is the SI unit of absorbed dose. One gray is equal to an absorbed dose of 1 joule per kilogram (100 rad).
9.2.2 Rad is the traditional unit of absorbed dose. One rad is equal to an absorbed dose of 100 erg per gram or 0.01 joule per kilogram. (0.01 Gy)
9.2.3 Rem is the traditional unit of any of the quantities expressed as dose equivalent. The dose equivalent in rem is equal to the absorbed dose in rad multiplied by the quality factor. (1 rem = 0.01 Sv)
9.2.4 Sievert is the SI unit of any of the quantities expressed as dose equivalent. The dose equivalent in sievert is equal to the absorbed dose in gray multiplied by the quality factor. (1 Sv = 100 rem)
9.3 As used in these regulations, the quality factors for converting absorbed dose to dose equivalent are shown in Table I:
Type of Radiation | Quality Factor (Q) | Absorbed Dose Equal to a Unit Dose Equivalenta/ |
X, gamma, or beta radiation and high-speed electrons | 1 | 1 |
Alpha particles, multiple-charged particles, fission fragments and heavy particles of unknown charge | 20 | 0.05 |
Neutrons of unknown energy | 10 | 0.1 |
High-energy protons | 10 | 0.1 |
a/ Absorbed dose in gray equal to 1 Sv or the absorbed dose in rad equal to 1 rem. | ||
9.4 If it is more convenient to measure the neutron fluence rate than to determine the neutron dose equivalent rate in sievert per hour or rem per hour, as provided in A.13c., 0.01 Sv (1 rem) of neutron radiation of unknown energies may, for purposes of these regulations, be assumed to result from a total fluence of 25 million neutrons per square centimeter incident upon the body. If sufficient information exists to estimate the approximate energy distribution of the neutrons, the licensee or registrant may use the fluence rate per unit dose equivalent or the appropriate Q value from Table II to convert a measured tissue dose in gray or rad to dose equivalent in sievert or rem.
Neutron Energy (MeV) | Quality Factora/ (Q) | Fluence per Unit Dose Equivalentb/ (Neutrons cm-2 rem -1) | Fluence per Unit Dose Equivalentb/ (Neutrons cm-2 Sv-1) | |
(thermal) | 2.5E-8 | 2 | 980E+6 | 980E+8 |
1E-7 | 2 | 980E+6 | 980E+8 | |
1E-6 | 2 | 810E+6 | 810E+8 | |
1E-5 | 2 | 810E+6 | 810E+8 | |
1E-4 | 2 | 840E+6 | 840E+8 | |
1E-3 | 2 | 980E+6 | 980E+8 | |
1E-2 | 2.5 | 1010E+6 | 1010E+8 | |
1E-1 | 7.5 | 170E+6 | 170E+8 | |
5E-1 | 11 | 39E+6 | 39E+8 | |
1 | 11 | 27E+6 | 27E+8 | |
2.5 | 9 | 29E+6 | 29E+8 | |
5 | 8 | 23E+6 | 23E+8 | |
7 | 7 | 24E+6 | 24E+8 | |
10 | 6.5 | 24E+6 | 24E+8 | |
14 | 7.5 | 17E+6 | 17E+8 | |
20 | 8 | 16E+6 | 16E+8 | |
40 | 7 | 14E+6 | 14E+8 | |
60 | 5.5 | 16E+6 | 16E+8 | |
1E+2 | 4 | 20E+6 | 20E+8 | |
2E+2 | 3.5 | 19E+6 | 19E+8 | |
3E+2 | 3.5 | 16E+6 | 16E+8 | |
4E+2 | 3.5 | 14E+6 | 14E+8 | |
a/ Value of quality factor (Q) at the point where the dose equivalent is maximum in a 30-centimeter diameter cylinder tissue-equivalent phantom. | ||||
b/ Monoenergetic neutrons incident normally on a 30-centimeter diameter cylinder tissue-equivalent phantom. | ||||
10.1 For purposes of these regulations, activity is expressed in the SI unit of becquerel (Bq) or in the special unit of curie (Ci), or their multiples, or disintegrations or transformations per unit of time.
10.2 One becquerel (Bq) = 1 disintegration or transformation per second (dps or tps).
10.3 One curie (Ci) = 3.7E+10 disintegrations or transformations per second (dps or tps) = 3.7E+10 becquerel (Bq) = 2.22E+12 disintegrations or transformations per minute (dpm or tpm).
Part B Registration of Radiation Source Facilities and Services
This Part provides for:
1.1 The registration of ionizing radiation source facilities, and
1.2 The registration of persons providing ionizing radiation source installation, servicing, and/or other services listed in this Part.
1.3 In addition to the requirements of this Part, all registrants are subject to the applicable provisions of the General Provisions (4465, Part A), Standards for Protection (4465, Part D), and Notices, Instructions and Reports (4465, Part J) and Compliance Procedures (4465, Part K). In addition, some registrants are subject to provisions of the regulations for Industrial Radiography (4465, Part E), Diagnostic X-Rays and Imaging Systems in the Healing Arts (4465, Part F), Analytical Equipment (4465, Part H) or Particle Accelerators (4465, Part I) and Therapeutic Radiation Machines (4465, Part X).
“Agency” means the Division of Public Health, Delaware Department of Health and Social Services.
“CFR” means Code of Federal Regulations.
“Chiropractic” means a drugless system of health care based on the principle that interference with the transmission of nerve impulses may cause disease, per 24 Del.C., Ch. 7, Board of Chiropractic, as amended.
“Certificate of Approval to Construct” means a document stipulating that work will be done in accordance to the plans and specifications as approved by the Office of Engineering. If at any point after the issuance of a certificate of Approval To Construct there are any changes made to the plans, the Office of Engineering must be immediately notified for them to take appropriate action.
“Certificate of Approval to Operate” means a document indicating that requirements for operation of a new radiation machine facility have been approved by the Office of Radiation Control, following a pre-operational, on-site inspection.
“Dentist” shall mean a person who is qualified to practice dentistry as prescribed in 24 Del.C., Ch. 11, Dentistry and Dental Hygiene, as amended.
“Facility” means the location, building, vehicle, or complex under one administrative control, at which one or more radiation sources are installed, located and/or used.
“Healing arts” includes but is not limited to the practice of medicine, surgery, dentistry, registered pharmacy, podiatry, osteopathy, chiropractic, veterinary medicine or nursing.
“kVP” or Peak Tube Potential, means the maximum value of the potential difference across the x-ray tube during an exposure. This value is usually included in manufacturer’s technical specification for an x-ray device.
“Licensed Practitioner” means a physician an individual licensed to practice medicine, dentistry, podiatry, chiropractic, osteopathy, or veterinary medicine in this state.
“Manager” means the individual working at the facility who is authorized by the owner to sign the application form as the applicant.
“Office of Engineering” means the office in the Delaware Division of Public Health which reviews radiation shielding plans, and issues approval for construction of new radiation machine facilities or rooms.
“Office of Radiation Control” means the office in the Delaware Division of Public Health which carries out the Delaware Radiation Control Regulations, issues radiation source facility registration permits, and performs on-site inspections of new and existing radiation machine facilities to determine compliance.
“Owner/Leasee” means the person/individual who owns/leases the radiation source. An out-of-state owner shall authorize a manager working at the facility to sign the application form.
"Physician" means an allopathic doctor of medicine and surgery or a doctor of osteopathic medicine and surgery who is registered and certified to practice medicine pursuant to 24 Del.C., Ch. 17, Medical Practice Act, as amended.
“Podiatrist” means a person who is qualified to practice podiatry and is licensed under 24 Del.C., Ch. 5, Podiatry, as amended.
“Principal Supervisor” means the Licensed Practitioner responsible for initiating use of x-ray equipment or other device generating ionizing radiation in the healing arts.
“Qualified Expert” means an individual who has satisfactorily fulfilled the training and experience requirements consistent with achieving a level of competency sufficient to function effectively in the position for which registration is sought. Such individuals must demonstrate to the satisfaction of the Agency their qualifications, for example, individuals certified in the appropriate field by the American Board of Radiology, or the American Board of Health Physics, or the American Board of Medical Physics, or those having equivalent qualifications. With reference to the calibration of radiation therapy equipment, an individual, in addition to the above qualifications, must be qualified in accordance with 4465 Part F and 4465 Part X of these regulations, as amended.
“Qualified Medical Physicist” means an individual qualified in accordance with 4465, Part X, Therapeutic Radiation Machines, of these regulations. who meets each of the following credentials:
1. Has earned a master's and/or doctoral degree in physics, medical physics, biophysics, radiological physics, medical health physics, or equivalent disciplines from an accredited college or university; and
2. Has been granted certification in the specific subfield(s) of medical physics with its associated medical health physics aspects by an appropriate national certifying body and abides by the certifying body's requirements for continuing education; and/or
3. Is credentialed in accordance with Part X, subsection 3.4, as amended.
“Radiation Source” see source of radiation.
“Radiation Safety Officer” or RSO for a radiation machine facility means an individual assigned to perform radiation safety duties who has training and experience in the safe and effective use of radiation machines, their potential radiation hazards, and emergency precautions applicable to the type of activity or facility to which the RSO is assigned.
“Radiation Service Provider” means company or person who provides radiation services to registered radiation source facilities in Delaware, see Section 9.0 of this Part.
“Source of Radiation” means any radioactive material or any device or equipment emitting, or capable of producing, ionizing radiation.
“Storage” means a condition in which a device or source is not being used for an extended period of time, and has been made inoperable and shall be tagged as out of service.
“Veterinarian” shall mean a person who has received a degree in veterinary medicine from a school of veterinary medicine, per 24 Del.C. Ch. 33, Veterinarians, as amended.
All registration permit-holders shall prohibit any person or company from furnishing radiation machine servicing or services to their radiation machine facility until such person provides evidence of registration with the Agency as a provider of services in accordance with Section, 9.0 of this Part.
4.1 Electronic equipment that produces radiation incidental to its operation for other purposes is exempt from the registration and notification requirements of this regulation, provided that the equivalent dose averaged over an area of 10 square centimeters does not exceed 5 µSv (0.5 millirem) per hour at 5 centimeters from any accessible surface of such equipment. The production, testing, or factory servicing of such equipment shall not be exempt.
4.2 Radiation machines in transit or in storage incident to transit are exempt from the requirements of this Part. This exemption does not apply to the providers of radiation machines for mobile services.
4.3 Domestic television receivers, computer monitors, and electron microscopes are exempt from the registration and notification requirements of this regulation.
5.1 Radiation machine facilities proposed for construction, renovation, or equipment installation after the effective date of this regulation that are designed to house x-ray machines with the potential to generate radiation dose to members of the public equal to or greater than 100 millirem per year, or expose a member of the public to an exposure rate equal to or greater than 2 milliroentgen per hour shall be required to submit a radiation shielding plan prepared by a Qualified Expert who is registered with the Office of Radiation Control as a Radiation Service Provider (see Section 9.0 of this part).
5.1.1 Radiation Machine Facilities or rooms which require a shielding plan include the following modalities:
5.2 New radiation machine facilities or rooms designed to house only x-ray machines that operate at maximum energy less than or equal to 70 kVP shall be exempt from the radiation shielding plan requirement; such devices include but are not limited to the following modalities:
5.2.1 Radiation Machine Facilities or rooms which generally do not require a shielding plan include the following modalities:
5.3 Prior to construction, the floor plans, shielding specifications and equipment arrangement of all new installations, or modifications of existing installations utilizing ionizing radiation sources with maximum energy greater than 70 kVP shall be submitted to the Division of Public Health Office of Engineering for review and approval. The required information is listed in Appendices A and B of this Part and Regulation 4465 Part X, Appendix A, for radiation therapeutic sources.
5.4 The Agency shall require the applicant to utilize the services of a Qualified Expert who is registered with the Agency [see Section 9.0 of this Part] to determine the shielding requirements prior to the plan review and approval. The registered consultant shall provide the shielding information on Form R15A or equivalent to the Office of Engineering, Division of Public Health, which will review the shielding plan and if determined acceptable, will issue a Certificate of Approval to Construct letter to the applicant.
5.5 The issuance of a Certificate of Approval to Construct by the Office of Engineering for radiation shielding plans shall not preclude the requirement of additional modifications should a subsequent analysis of operating conditions indicate the possibility of an individual receiving a dose in excess of the limits prescribed in Regulations 4465 Part D, (Sections D.201, 207, 208 and 301) of these regulations.
5.6 The Office of Radiation Control, Division of Public Health shall conduct a pre-operational, on-site inspection to evaluate shielding and/or operating conditions prior to issuance of the radiation machine registration permit and Certificate of Approval to Operate.
5.7 The Certificate of Approval to Operate issued by the Office of Radiation Control reflects regulatory compliance at the time of the pre-operational inspection of a new facility, and does not imply or certify the facility beyond the scope of that specific inspection.
5.8 After installation of any radiation machine, the registrant shall maintain for inspection by the Agency:
5.8.1 The maximum rated technique factors of each machine;
5.8.2 A scale drawing of the room in which a stationary radiation machine system is located with such drawing indicating the use of areas adjacent to the room and an estimation of the extent of occupancy by an individual in such areas. In addition, the drawing shall include:
5.8.2.1 The results of a survey for radiation levels present at the operator's position and at pertinent points outside the room at specified test conditions; or
5.8.2.2 The type and thickness of materials, or lead equivalency, of each protective barrier.
5.9 Radiation machine facilities that initiated design, construction or installation of dental panoramic, cephalometric, or cone beam CT devices prior to the effective date of this regulation shall maintain records of radiation surveys of exposure rate (milliroentgen per hour) levels present in uncontrolled public areas (ie. corridors or alcoves) where members of the public or employees may be present in the facility. If such surveys indicate an exposure rate equal to or greater than 2 millroentgen per hour is possible in uncontrolled public areas while x-ray equipment is in operation the facility shall provide administrative controls to limit the dose to members of the public with a visible barrier to delineate the controlled area.
6.1 Each owner of a radioactive material facility shall:
6.1.1 Apply for registration of such facility with the Agency prior to the receipt, possession, use, sale, transfer, ownership or acquisition of the radioactive material. Application for registration shall be completed on forms furnished by the Agency.
6.1.2 Designate on the application form an individual to be responsible for radiation protection, duties; (Radiation Safety Officer), address of the facility, and for the radioactive material; element name, atomic mass, chemical or physical form and maximum amount to be possessed at any one time.
6.2 Each owner of a radiation machine facility shall:
6.2.1 Apply for registration of such facility with the Agency prior to the operation of a radiation source facility. Application for registration shall be completed on forms furnished by the Agency and shall contain all the information required by the form and accompanying instructions;
6.2.2 Designate on the application form an individual to be responsible for radiation protection duties; (Radiation Safety Officer); per Appendix C of this Part.
6.2.3 A Licensed Practitioner responsible for directing the operation of radiation machines shall be designated on each healing arts x-ray facility application, specifying their Delaware license number and phone number. The signature of the administrator, president, or chief executive officer will be accepted in lieu of a licensed practitioner's signature if the facility has more than one licensed practitioner (for example, hospitals, large clinics, or multi-practitioner practices), except where prohibited by State Law.
6.2.4 Prohibit any person from furnishing radiation source servicing or services as described in section. 9.4 of this Part to their radiation source facility, until such person provides evidence that they have been registered with the Agency as a Radiation Service Provider in accordance with section 9.0 of this part.
6.2.5 In any facility regulated by or requiring registration under these regulations, the registration permit-holder shall allow only individuals who are adequately trained in radiation safety and the safe and effective use of the machine to operate any radiation machine.
6.2.5.1 The facility registration permit-holder shall document evaluation of the qualifications of each individual permitted to operate any radiation machine at the facility.
6.2.5.1.1 Each operator shall meet all radiation safety training and experience requirements of the respective State of Delaware professional licensure board, as applicable, and any applicable requirements of these regulations (4465 Part B), and 4466 Radiation Technologist/Technician Certification Regulations.
7.1 In addition to the requirements of Section 6.0 of this Part, the applicant shall submit the following information:
7.1.1 An established main location where the machine(s), records, etc. will be maintained for inspection. This shall be a street address, not a post office box number.
7.1.2 A sketch or description of the normal configuration of each radiation machine's use, including the operator's position and any ancillary personnel's location during exposures. If a mobile van is used with a fixed unit inside, furnish the floor plan indicating protective shielding and the operator's location; and
7.1.3 A current copy of the applicant's operating and safety procedures including radiological practices for protection of patients, operators, employees, and the general public.
8.1 In addition to the requirements of 6.0 of this Part each applicant shall apply for and receive authorization for healing arts screening before initiating a screening program. The information and evaluation in Appendix E of this part shall be submitted with the application.
8.2 In addition to the requirements of 6.0 of this Part, any research using radiation machines on humans shall be approved by an Institutional Review Board (IRB) as required by Title 45, CFR, Part 46 and Title 21, CFR, Part 56, as amended.
9.1 Each person or company who is engaged in the business of installing or offering to install radiation machines or is engaged in the business of furnishing or offering to furnish radiation machine servicing or services in this State shall apply for registration of such services with the Agency, and receive Agency approval prior to furnishing or offering to furnish any such services.
9.2 Application for registration shall be completed on forms furnished by the Agency and shall contain all information required by the Agency as indicated on the forms and accompanying instructions.
9.3 Each Radiation Service Provider applying for registration under this regulation shall specify:
9.3.1 That they have read and understand the requirements of this and other applicable regulations;
9.3.2 The education and training that qualify them to discharge the services for which they are applying for registration.
9.4 For the purpose of section 9.0, services may include but shall not be limited to:
9.4.1 Installation and/or servicing of radiation sources and associated radiation source components;
9.4.2 Calibration of radiation source or radiation measurement instruments or devices;
9.4.3 Radiation protection or health physics consultations or surveys;
9.4.4 Personnel dosimetry services;
9.4.5 Radiation Shielding Plans for X-Ray Rooms; or
9.4.6 Practice as a Qualified Medical Physicist.
9.5 No individual working as a Radiation Service Provider shall perform services which are not specifically authorized for that individual by the Agency.
10.1 Upon a determination that an applicant meets the requirements of the regulations, the Agency shall issue a notice of registration, which shall be displayed by the registrant in public view.
10.2 The Agency may incorporate in the notice of registration at the time of issuance or thereafter by appropriate rule, regulation, or order, such additional requirements and conditions with respect to the registrant's receipt, possession, use, sale and/or transfer of ownership responsibility of radiation sources as it deems appropriate or necessary.
Except as provided in Section 12.0 below, each notice of registration shall expire at the end of the specified day in the month and year stated therein.
12.1 Application for renewal of registration shall be filed in accordance with Part B, sSections 6.0, 7.0 and/or 9.0 of this Part.
12.2 In any case in which a registrant not less than 30 days prior to the expiration of his existing notice of registration has filed an application in proper form for renewal, such existing notice of registration shall not expire until the application status has been finally determined by the Agency.
The registrant shall notify the Agency in writing on forms furnished by the Agency before making any change which would render the information contained in the application for registration and/or the notice of registration no longer accurate. The Agency shall incorporate such changes and issue a corrected registration if necessary.
No person, in any advertisement, shall refer to the fact that he or his facility is registered with the Agency pursuant to the provisions of Part B, sections Section 6.0 or 9.0, and no person shall state or imply that any activity under such registration has been approved by the Agency.
15.1 Any person who sells, leases, transfers, lends, disposes, assembles, or installs radiation sources in this State shall notify the Agency within 15 days of:
15.1.1 The name and address of persons who have received these sources;
15.1.2 The manufacturer, model, and serial number of each radiation source transferred; and
15.1.3 The date of transfer of each radiation source.
15.1.4 In the case of diagnostic x-ray systems which contain certified components, a copy of the assembler's report prepared in compliance with requirements of the Federal diagnostic x-ray standard (21 CFR 1020.30(d)) shall be submitted to the Agency following completion of the assembly. Such report shall suffice in lieu of any other report by the assembler.
15.2 No person shall make, sell, lease, transfer, lend, assemble, or install radiation sources or the supplies used in connection with such machines unless such supplies and equipment when properly placed in operation and used shall meet the requirements of these regulations.
16.1 Whenever any radiation source is to be brought into the State, for any temporary use, the person proposing to bring such source into the State shall submit a complete, prescribed application form to the Agency and must receive Agency approval at least 2 working days before such machine is to be brought into the State. The applicant must receive Agency approval prior to use. The notice shall include:
16.1.1 The number(s) and type(s) of radiation source(s);
16.1.2 The nature, start date, duration, and scope of use;
16.1.3 The exact location(s) where the radiation source is to be used; and
16.1.4 the name(s) of the Delaware licensed practitioner(s) and their professional license number(s) if the sources are used to irradiate human beings;
16.1.5 a copy of the person's home state registration license or equivalent document; and
16.1.6 the name(s) and address(es) where the source user(s) can be reached while in the state.
16.2 The person proposing to bring such out-of-state source into Delaware referred to in section 16.1 shall:
16.2.1 Comply with all applicable regulations of the Agency;
16.2.2 Supply the Agency with such other information as the Agency may reasonably request; and
16.2.3 Not operate within the state on a temporary basis in excess of 90 days. Permission to operate for more than 90 days may be granted by the Agency up to 180 days per year.
In order for the Agency to provide an evaluation, technical advice, and official approval on shielding requirements for a radiation installation, the following information must be submitted to the Office of Engineering in the Division of Public Health. The Agency may require a pre-operational inspection be conducted by the Office of Radiation Control to assure that design and operational safety requirements are met prior to approval of the radiation machine registration permit.
1. The plans showing, as a minimum, the following:
(a) The normal location of the system's radiation port; the port's travel and traverse limits; general direction(s) of the useful beam; locations of any windows and doors or other openings; the location of the operator's booth; and the location of the control panel;
(b) The structural composition and thickness or lead equivalence of all walls, doors, partitions, floor, and ceiling of the room(s) concerned;
(c) The dimensions of the room(s) concerned;
(d) The type of occupancy of all adjacent areas inclusive of space above and below the room(s) concerned. If there is an exterior wall, show distance to the closest area(s) where it is likely that individuals may be present;
(e) The make and model of the equipment, the maximum technique factors, and the energy waveform (single phase, three phase, etc.);
(f) The type of examination(s) or treatment(s) which will be performed with the equipment.
2. Information on the anticipated workload of the system(s) in mA-minutes per week.
3. A report showing all basic assumptions used in the development of the shielding specifications.
1. Space Requirements:
(a) The operator shall be allotted not less than 0.70 m2 (7.5 square feet) of unobstructed floor space in the booth;
(b) The operator's booth may be any geometric configuration with no dimension of less than 0.6 m (2 feet);
(c) The space shall be allotted excluding any encumbrance by the x‑ray control panel, such as overhang, cables, or other similar encroachments;
(d) The booth shall be located or constructed such that unattenuated direct scatter radiation originating on the examination table or at the wall-mounted image receptor will not reach the operator's position in the booth.
2. Structural Requirements:
(a) The booth walls shall be permanently fixed barriers of at least 2 m (7 feet) high;
(b) When a door or movable panel is used as an integral part of the booth structure, it must have an interlock which will prevent an exposure when the door or panel is not closed;
(c) Shielding shall be provided to meet the requirements of Part D of these regulations.
3. Radiation Exposure Control Placement:
The radiation exposure control for the system shall be fixed within the booth and:
(a) Shall be at least 1.0 m (40 inches) from any point subject to direct scatter, leakage or primary beam radiation;
(b) Shall allow the operator to use the majority of the available viewing windows.
4. Viewing System Requirements:
(a) Each booth shall have at least one viewing device which will:
(1) Be so placed that the operator can view the patient during any exposure; and
(2) Be so placed that the operator can have full view of any occupant of the room and should be so placed that the operator can view any entry into the room. If any door which allows access to the room cannot be seen from the booth, then outside that door there shall be an "x-ray on" warning sign that will be lighted anytime the rotor of the x-ray tube is activated. Alternatively, an interlock shall be present such that exposures are prevented unless the door is closed.
(b) When the viewing system is a window, the following requirements also apply:
(1) The window shall have a viewing area of at least 0.09 m2 (1 square foot);
(2) Regardless of size or shape, at least 0.09 m2 (1 square foot) of the window area must be centered no less than 0.6 m (2 feet) from the open edge of the booth and no less than 1.5 m (5.0 feet) from the floor;
(3) The window shall have at least the same lead equivalence as that required in the booth's wall in which it is mounted.
(c) When the viewing system is by mirrors, the mirror(s) shall be so located as to accomplish the general requirements of Appendix B4.(a).
(d) When the viewing system is by electronic means:
(1) The camera shall be so located as to accomplish the general requirements of Appendix B4.(a); and
(2) There shall be an alternate viewing system as a backup for the primary system.
The applicant or registration permit-holder, shall require each individual assigned to fulfill responsibilities and duties as Radiation Safety Officer (RSO) to be an individual who has training and experience in the safe and effective use of radiation machines and the potential radiation hazards and emergency precautions applicable to the type(s) of activity or facility for which the individual is seeking to perform RSO duties, to include:
I. Establishing and overseeing operating and safety procedures that maintain radiation exposures as low as reasonably achievable (ALARA), and to review them periodically to ensure that the procedures are current and conform with these regulations;
II. Ensuring that individual monitoring devices are properly used by occupationally exposed personnel as required by the regulations, that records are kept of the monitoring results, and that timely notifications are made as required by 4465 Part D;
III. Investigating and reporting to the agency each known or suspected case of radiation exposure to an individual or radiation level detected in excess of limits established by these regulations and each theft or loss of source(s) of radiation, determining the cause, and taking steps to prevent its recurrence;
IV. Having a thorough knowledge of management policies, administrative procedures and records of the registration permit-holder and keeping management informed on a periodic basis of the performance of the registrant’s radiation protection program, if applicable;
V. Assuming control and having the authority to institute corrective actions including shutdown of operations when necessary in emergency situations or unsafe conditions;
VI. Maintaining records as required by these regulations; and
VII. Ensuring that personnel are adequately trained and complying with these regulations, the conditions of the certificate of registration, and the operating and safety procedures of the registered permit-holder.
All persons performing radiation machine assembly, installation or repair shall meet the general requirements in subparagraph 1. of this paragraph and one or more of the specialized requirements in subparagraph 2. of this paragraph.
1. General requirements include:
(a) Experience or education providing familiarity with the type(s) of equipment to be serviced, to include radiation safety;
(b) Knowledge of protective measures to reduce potentially hazardous conditions; and
(c) Six months of supervised assembly and repair of the type(s) of equipment to be serviced.
2. Specialized requirements include:
(a) One year of formal training (may be satisfied by factory school, military technical training school, or other courses in radiation machine assembly, installation or repair techniques) or an associate's degree in biomedical equipment repair;
(b) A bachelor's degree in electrical engineering with specialized training in radiation producing devices; or
(c) A combination of training and experience equal to clause (a) of this subparagraph.
The following information must be submitted by persons proposing to conduct healing arts screening. Persons requesting that the agency approve a healing arts screening program shall submit the following information and evaluation.
1. Administrative controls to include the following:
(a) The name and address of the applicant and, where applicable, the names and addresses of agents within the state;
(b) The diseases or conditions for which the x‑ray examinations are to be used in diagnoses;
(c) A detailed description of the x‑ray examinations proposed in the screening program;
(d) A description of the population to be examined in the screening program, for example, age, sex, physical condition, and other appropriate information;
(e) An evaluation of any known alternate methods not involving ionizing radiation that could achieve the goals of the screening program and why these methods are not used instead of the x‑ray examination; and
(f) For mobile screening operations, location(s) where radiation machines are used and maintained.
2. Operating procedures for all x-ray systems (except bone densitometers) to include the following:
(a) An evaluation of the x-ray systems to be used in the screening program. The evaluation shall be performed by a licensed medical physicist with a specialty in diagnostic radiological physics. The evaluation shall show that such systems do satisfy all requirements of this section;
(b) A description of the diagnostic imaging quality control program; and
(c) A copy of the technique chart for the x‑ray examination procedures to be used.
3. Operating procedures for bone densitometers to include the manufacturer's evaluation of the system to be used in the screening program. The evaluation shall show that such systems satisfy all requirements of this section.
4. Training data to include the following:
(a) The qualifications of each individual who will be operating the x‑ray systems;
(b) The qualifications of the individual who will be supervising the operators of the x‑ray systems. The extent of supervision and the method of work performance evaluation shall be specified; and
(c) The name and address of the practitioner licensed in the state who will interpret the radiographs.
5. Records to include the following:
(a) A description of the procedures to be used in advising the individuals screened, and their private practitioners of the healing arts, of the results of the screening procedure and any further medical needs indicated; and
(b) A description of the procedures for the retention or disposition of the radiographs and other records pertaining to the x‑ray examinations.
This part provides for the licensing of radioactive material, for purposes of protecting the public health and safety. No person shall receive, possess, use, transfer, sell, own or acquire radioactive material except as authorized in a specific or general license per the U.S. Nuclear Regulatory Commission (NRC), in accordance with Title 10 – Code of Federal Regulations. Primary radioactive material licensing and enforcement authority was transferred to the NRC in 2007, pursuant to the Federal Energy Policy Act of 2005. However, radioactive material facilities must be registered with the State of Delaware in accordance with 4481/Part B of these regulations.
STANDARDS FOR PROTECTION AGAINST RADIATION
General Provisions
1.1 Part D establishes standards for protection against ionizing radiation resulting from activities conducted pursuant to licenses or registrations issued by the Agency. These regulations are issued pursuant to the Title 16, Delaware Code, Chapter 74 Radiation Control.
1.2 The requirements of Part D are designed to control the receipt, possession, use, sale, transfer, and disposal of sources of radiation by any licensee or registrant so the total dose to an individual, including doses resulting from all sources of radiation other than background radiation, does not exceed the standards for protection against radiation prescribed in Part D. However, nothing in Part D shall be construed as limiting actions that may be necessary to protect health and safety.
Except as specifically provided in other Parts of these regulations, Part D applies to persons licensed or registered by the Agency to receive, possess, use, sell, transfer, or dispose of sources of radiation. The limits in Part D do not apply to doses due to background radiation, to exposure of patients to radiation for the purpose of medical diagnosis or therapy, to exposure from individuals administered radioactive material and released in accordance with the providers ALARA license conditions, or to voluntary participation in medical research programs.
As used in Part D:
"A1" means the maximum activity of special form radioactive material permitted in a Type A package.
"A2" means the maximum activity of radioactive material, other than special form radioactive material, permitted in a Type A package.
These values are either listed in Appendix A of Part T of these regulations, Table I, or may be derived in accordance with the procedure prescribed in Appendix A of Part T of these regulations.
“Absorbed dose” means the energy imparted by ionizing radiation per unit mass of irradiated material. The units of absorbed dose are the gray (Gy) and the rad.
“Accelerator” means any machine capable of accelerating electrons, protons, deuterons, or other charged particles in a vacuum and of discharging the resultant particulate or other radiation into a medium at energies usually in excess of 1 MeV. For purposes of this definition, "particle accelerator" is an equivalent term.
“Accelerator-produced material” means any material made radioactive by a particle accelerator.
“Activity” means the rate of disintegration or transformation or decay of radioactive material. The units of activity are the becquerel (Bq) and the curie (Ci).
“Address of use” means the building or buildings that are identified on the permit (license) and where radioactive materials may be produced, prepared, received, used, or stored.
"Adult" means an individual 18 or more years of age.
“Agency” means the Division of Public Health, Delaware Department of Health and Social Services.
“Agreement State” means any State with which the Nuclear Regulatory Commission or the Atomic Energy Commission has entered into an effective agreement under subsection 274b. of the Atomic Energy Act of 1954, as amended (73 Stat. 689).
“Airborne radioactive material” means any radioactive material dispersed in the air in the form of dusts, fumes, particulates, mists, vapors, or gases.
“Airborne radioactivity area” means a room, enclosure, or area in which airborne radioactive materials exist in concentrations:
(1) In excess of the derived air concentrations (DAC's) specified in Appendix B, Table I of Part D of these regulations; or
(2) To such a degree that an individual present in the area without respiratory protective equipment could exceed, during the hours an individual is present in a week, an intake of 0.6 percent of the annual limit on intake (ALI) or 12 DAC-hours.
“Airline respirator” (see "Supplied-air respirator (SAR)").
“Air-purifying respirator” means a respirator with an air-purifying filter, cartridge, or canister that removes specific air contaminants by passing ambient air through the air-purifying element.
“Annual limit on intake (ALI)” means the derived limit for the amount of radioactive material taken into the body of an adult worker by inhalation or ingestion in a year. ALI is the smaller value of intake of a given radionuclide in a year by the reference man that would result in a committed effective dose equivalent of 5 rems (0.05 Sv) or a committed dose equivalent to 50 rems (0.5 Sv) to any individual organ or tissue. (ALI values for intake by ingestion and by inhalation of selected radionuclides are given in Appendix B of this regulation.
“As low as is reasonably achievable” (ALARA) means making every reasonable effort to maintain exposures to radiation as far below the dose limits in these regulations as is practical, consistent with the purpose for which the licensed or registered activity is undertaken, taking into account the state of technology, the economics of improvements in relation to state of technology, the economics of improvements in relation to benefits to the public health and safety, and other societal and socioeconomic considerations, and in relation to utilization of nuclear energy and licensed or registered sources of radiation in the public interest.
“Assigned Protection Factor (APF)” means the expected workplace level of respiratory protection that would be provided by a properly functioning respirator or a class of respirators to properly trained and fitted users. Operationally, the inhaled concentration can be estimated by dividing the ambient airborne concentration by the APF.
“Atmosphere-supplying respirator” means a respirator that supplies the respirator user with breathing air from a source independent of the ambient atmosphere, and includes supplied-air respirators (SAR’s) and self-contained breathing apparatus (SCBA) units.
“Authorized user” means a practitioner of the healing arts who is identified as an authorized user on an Agency, Agreement State, Licensing State or the Nuclear Regulatory Commission license that authorizes the medical use of radioactive material.
“Background radiation” means radiation from cosmic sources, naturally occurring radioactive material, (which has not been technologically enhanced) including radon, except as a decay product of source or special nuclear material, and including global fallout as it exists in the environment from the testing of nuclear explosive devices, or from past nuclear accidents such as Chernobyl that contribute to background radiation and are not under the control of the licensee or registrant. "Background radiation" does not include sources of radiation from radioactive materials regulated by the Agency.
"Becquerel" (Bq) means the Standard Internationale (SI) unit of activity. One becquerel is equal to 1 disintegration or transformation per second (dps or tps).
"Bioassay" means the determination of kinds, quantities or concentrations, and, in some cases, the locations of radioactive material in the human body, whether by direct measurement, in vivo counting, or by analysis and evaluation of materials excreted or removed from the human body. For purposes of these regulations, "radiobioassay" is an equivalent term.
"Brachytherapy" means a method of radiation therapy in which radiation sources are utilized to deliver a radiation dose at a distance of up to a few centimeters, by surface, intracavitary, intraluminal, or interstitial application.
"Byproduct material" means:
(1) Any radioactive material (except special nuclear material) yielded in, or made radioactive by, exposure to the radiation incident to the process of producing or using special nuclear material;
(2) The tailings or wastes produced by the extraction or concentration of uranium or thorium from ore processed primarily for its source material content, including discrete surface wastes resulting from uranium solution extraction processes. Underground ore bodies depleted by these solution extraction operations do not constitute "byproduct material" within this definition;
(3) (i) Any discrete source of radium-226 that is produced, extracted, or converted after extraction, before, on, or after August 8, 2005, for use for a commercial, medical, or research activity; or
(ii) Any material that—
(A) Has been made radioactive by use of a particle accelerator; and
(B) Is produced, extracted, or converted after extraction, before, on, or after August 8, 2005, for use for a commercial, medical, or research activity; and
(4) Any discrete source of naturally occurring radioactive material, other than source material, that—
(i) The Commission, in consultation with the Administrator of the Environmental Protection Agency, the Secretary of Energy, the Secretary of Homeland Security, and the head of any other appropriate Federal agency, determines would pose a threat similar to the threat posed by a discrete source of radium-226 to the public health and safety or the common defense and security; and
(ii) Before, on, or after August 8, 2005, is extracted or converted after extraction for use in a commercial, medical, or research activity.
“Calendar quarter” means not less than 12 consecutive weeks nor more than 14 consecutive weeks. The first calendar quarter of each year shall begin in January and subsequent calendar quarters shall be so arranged such that no day is included in more than one calendar quarter and no day in any one year is omitted from inclusion within a calendar quarter. The method observed by the licensee or registrant for determining calendar quarters shall only be changed at the beginning of a year.
“Calibration” means the determination of (1) the response or reading of an instrument relative to a series of known radiation values over the range of the instrument, or (2) the strength of a source of radiation relative to a standard.
“CFR” means Code of Federal Regulations.
“Chiropractic” means a drugless system of health care based on the principle that interference with the transmission of nerve impulses may cause disease, per Title 24 Delaware Code, Chapter 7, Board of Chiropractic, as amended.
“Class (or lung class or inhalation class)” means a classification scheme for inhaled material according to its rate of clearance from the pulmonary region of the lung. Materials are classified as D, W, or Y, which applies to a range of clearance half-times: for Class D (Days) of less than 10 days, for Class W (Weeks) from 10 to 100 days, and for Class Y (Years) of greater than 100 days.
“Collective dose” means the sum of the individual doses received in a given period of time by a specified population from exposure to a specified source of radiation.
“Committed dose equivalent” (HT.50) means the dose equivalent to organs or tissues of reference (T) that will be received from an intake of radioactive material by an individual during the 50-year period following the intake.
“Committed effective dose equivalent” (HE.50) is the sum of the products of the weighting factors (wT) applicable to each of the body organs or tissues that are irradiated and the committed dose equivalent to each of these organs or tissues (HE,50 = Σ wT HT,50).
“Constraint (dose constraint)” means a value above which specified licensee actions are required.
“Controlled area” means an area, outside of a restricted but inside the site boundary, access to which can be limited by the licensee or registrant, for any reason.
“Critical group” means the group of individuals reasonably expected to receive the greatest exposure to residual radioactivity for any applicable set of circumstances.
“Curie” means the traditional unit of quantity of activity. One curie (Ci) is that quantity of radioactive material, which decays at the rate of 3.7E+10 disintegrations or transformations per second (dps or tps).
“Declared pregnant woman” means a woman who has voluntarily informed the licensee, in writing, of her pregnancy and the estimated date of conception. The declaration remains in effect until the declared pregnant woman withdraws the declaration in writing or is no longer pregnant.
“Deep dose equivalent” (Hd), which applies to external whole body exposure, means the dose equivalent at a tissue depth of 1 centimeter (1000 mg/cm2).
“Demand respirator” means an atmosphere-supplying respirator that admits breathing air to the face piece only when a negative pressure is created inside the facepiece by inhalation
“Dentist” shall mean a person who is qualified to practice dentistry as prescribed in Title 24 Delaware Code, Chapter 11, Dentistry and Dental Hygiene, as amended.
“Department of Energy” means the Department of Energy established by Public Law 95-91, August 4, 1977, 91 Stat. 565, 42 U.S.C. Section 7101 as amended et seq., to the extent that the Department exercises functions formerly vested in the Atomic Energy Commission, its Chairman, members, officers and components and transferred to the Energy Research and Development Administration and to the Administrator thereof pursuant to sections 104(b), (c) and (d) of the Energy Reorganization Act of 1974 (Public Law 93-438, October 11, 1974, 88 Stat. 1233 at 1237, 42 U.S.C. 5814, effective January 19, 1975) and re-transferred to the Secretary of Energy pursuant to section 301(a) of the Department of Energy Organization Act (Public Law 95-91, August 4, 1977, 91 Stat. 565 at 577-578, 42 U.S.C. 7151, effective October 1, 1977 as amended.)
"Depleted uranium" means the source material uranium in which the isotope uranium-235 is less than 0.711 weight percent of the total uranium present. Depleted uranium does not include special nuclear material.
“Derived air concentration (DAC)” means the concentration of a given radionuclide in air which, if breathed by the reference man for a working year of 2,000 hours under conditions of light work (inhalation rate 1.2 cubic meters of air per hour), results in an intake of one ALI. DAC values are given in Appendix B of this regulation.
“Discrete Source” means a radionuclide that has been processed so that its concentration within a material has been purposely increased for use for commercial, medical, or research activities.
"Disposable respirator" means a respirator for which maintenance is not intended and that is designed to be discarded after excessive breathing resistance, sorbent exhaustion, physical damage, or end-of-service-life renders it unsuitable for use. Examples of this type of respirator are a disposable half-mask respirator or a disposable escape-only self-contained breathing apparatus (SCBA).
"Distinguishable from background" means that the detectable concentration of a radionuclide is statistically different from the background concentration of that radionuclide in the vicinity of the site or, in the case of structures, in similar materials using adequate measurement technology, survey, and statistical techniques.
"Dose" is a generic term that means absorbed dose, dose equivalent, effective dose equivalent, committed dose equivalent, committed effective dose equivalent, total organ dose equivalent, or total effective dose equivalent. For purposes of these regulations, "radiation dose" is an equivalent term.
"Dose equivalent (HT)" means the product of the absorbed dose in tissue, quality factor, and all other necessary modifying factors at the location of interest. The units of dose equivalent are the sievert (Sv) and rem.
"Dose limits" means the permissible upper bounds of radiation doses established in accordance with these regulations. For purposes of these regulations, "limits" is an equivalent term.
"Effective dose equivalent (HE)" means the sum of the products of the dose equivalent to the organ or tissue (HT) and the weighting factor (wT) applicable to each of the body organs or tissues that are irradiated (HE = Σ wTHT).
"Embryo/fetus" means the developing human organism from conception until the time of birth.
"Exposure" generally means being exposed to ionizing radiation or to radioactive material;
"Exposure Units" specifically as used in these regulations, the SI unit of exposure is coulomb per kilogram (C/kg), see Section 9.1 of Part A for Units of Exposure and Dose.
"Exposure rate" means the exposure per unit of time, such as roentgen per minute or milliroentgen per hour.
"External dose" means that portion of the dose equivalent received from any source of radiation outside the body.
“External Source” means all ionizing radiation sources that could present exposure or external dose to an individual.
"Extremity" means hand, elbow, and arm below the elbow, foot, knee, and leg below the knee.
“Facility” means the location, building vehicle, or complex under one administrative control, at which one or more radiation sources are installed, located and/or used.
"Filtering facepiece (dust mask)" means a negative pressure particulate respirator with a filter as an integral part of the facepiece or with the entire facepiece composed of the filtering medium, not equipped with elastomeric sealing surfaces and adjustable straps.
"Fit factor" means a quantitative estimate of the fit of a particular respirator to a specific individual, and typically estimates the ratio of the concentration of a substance in ambient air to its concentration inside the respirator when worn.
"Fit Test" means the use of a protocol to qualitatively evaluate the fit of a respirator on an individual.
"Former Atomic Energy Commission or Nuclear Regulatory Commission licensed facilities" means nuclear reactors, nuclear fuel reprocessing plants, uranium enrichment plants, or critical mass experimental facilities where Atomic Energy Commission or Nuclear Regulatory Commission licenses have been terminated.
"Generally applicable environmental radiation standards" means standards issued by the Environmental Protection Agency under the authority of the Atomic Energy Act of 1954, as amended, that impose limits on radiation exposures or levels, or concentrations or quantities of radioactive material, in the general environment outside the boundaries of locations under the control of persons possessing or using radioactive material.
"Gray" (Gy) means the Standard Internationale (SI) unit of absorbed dose. One gray is equal to an absorbed dose of 1 joule per kilogram (100 rad).
"Hazardous waste" means those wastes designated as hazardous by the Environmental Protection Agency regulations in 40 CFR Part 261, as amended.
“Healing arts” includes but is not limited to the practice of medicine, surgery, dentistry, registered pharmacy, podiatry, osteopathy, chiropractic, or veterinary medicine or nursing.
“Healing arts screening” means the testing of human beings using x-ray machines for the detection or evaluation of health indications when such tests are not specifically and individually ordered by a licensed practitioner of the healing arts legally authorized to prescribe such x-ray tests for the purpose of diagnosis or treatment.
"Helmet" means a rigid respiratory inlet covering that also provides head protection against impact and penetration.
"High radiation area" means an area, accessible to individuals, in which radiation levels from radiation sources external to the body could result in an individual receiving a dose equivalent in excess of 1 mSv (0.1 rem) in 1 hour at 30 centimeters from any source of radiation or 30 centimeters from any surface that the radiation penetrates.
"Hood" means a respiratory inlet covering that completely covers the head and neck and may also cover portions of the shoulders and torso.
"Human use" means the internal or external administration of radiation or radioactive material to human beings.
"Individual" means any human being.
"Individual monitoring" means the assessment of:
(1) Dose equivalent (a) by the use of individual monitoring devices or (b) by the use of survey data; or
(2) Committed effective dose equivalent (a) by bioassay or (b) by determination of the time-weighted air concentrations to which an individual has been exposed, that is, DAC-hours. [See the definition of DAC-hours in 4465 Part D of these regulations.]
(3) Dose equivalent by the use of survey data.
"Individual monitoring devices" means devices designed to be worn by a single individual for the assessment of dose equivalent. For purposes of these regulations, "personnel dosimeter" and "dosimeter" are equivalent terms. Examples of individual monitoring devices are film badges, thermoluminescence dosimeters (TLDs), pocket ionization chambers, optically stimulated luminescence (OSL) dosimeters and personal (lapel) air sampling devices.
"Inspection" means an official examination or observation including, but not limited to, tests, surveys, and monitoring to determine compliance with rules, regulations, orders, requirements, and conditions of the Agency.
"Instrument traceability" (for ionizing radiation measurements) means the ability to show that an instrument has been calibrated at specified time intervals using a national standard or a transfer standard. If a transfer standard is used, the calibration must be at a laboratory accredited by a program, which requires continuing participation in measurement quality assurance with the National Institute of Standards and Technology, or other equivalent national or international program.
"Interlock" means a device arranged or connected such that the occurrence of an event or condition is required before a second event or condition can occur or continue to occur.
"Internal dose" means that portion of the dose equivalent received from radioactive material taken into the body.
"JRCECT" means Joint Review Committee on Education in Cardiovascular Technology
“JRCNMT” means Joint Review Committee on Nuclear Medicine Technology
"JRCERT" means Joint Review Committee on Education in Radiologic Technology
"Lens dose equivalent (LDE)" means the external exposure to the lens of the eye as the dose equivalent at a tissue depth of 0.3 centimeter (300 mg/cm2).
"License" means a license issued by the US Nuclear Regulatory Commission, Agreement State, or the Agency, in accordance with applicable federal or state regulations, as amended.
"Licensed [or registered] material" means radioactive material received, possessed, used, transferred or disposed of under a general or specific license [or registration] issued by the Agency.
“Licensed Practitioner” means an individual licensed to practice medicine, dentistry, podiatry, chiropractic, osteopathy, advanced practice nursing, or veterinary medicine in this state.
"Licensee" means the holder of a license.
"Limits" [See "Dose limits"].
"Loose-fitting facepiece" means a respiratory inlet covering that is designed to form a partial seal with the face.
"Lost or missing source of radiation" means licensed [or registered] source of radiation whose location is unknown. This definition includes, but is not limited to, radioactive material that has been shipped but has not reached its planned destination and whose location cannot be readily traced in the transportation system.
"Major processor" means a user processing, handling, or manufacturing radioactive material exceeding Type A quantities as unsealed sources or material, or exceeding 4 times Type B quantities as sealed sources, but does not include nuclear medicine programs, universities, industrial radiographers, or small industrial programs. Type A and B quantities are defined in T.2 of these regulations.
"Member of the public" means any individual except when that individual is receiving an occupational dose.
"Minor" means an individual less than 18 years of age.
“Misadministration” means an event that meets the criteria in 4465 Part X, Therapeutic Radiation Machines, Section 5.2 of these regulations.
"Monitoring" means the measurement of radiation, radioactive material concentrations, surface area activities or quantities of radioactive material and the use of the results of these measurements to evaluate potential exposures and doses. For purposes of these regulations, "radiation monitoring" and "radiation protection monitoring" are equivalent terms.
"Natural radioactivity" means radioactivity of naturally occurring nuclides.
"Negative pressure respirator (tight fitting)" means a respirator in which the air pressure inside the facepiece is negative during inhalation with respect to the ambient air pressure outside the respirator.
"NORM" means any naturally occurring radioactive material. It does not include byproduct, source, or special nuclear material.
“Notice of Violation” means a written statement of one or more alleged infringements of a legally binding requirement. The notice normally requires the licensee, registrant or other permit holder to provide a written statement describing the following:
Corrective steps taken by the licensee, registrant or other permit holder and the results achieved;
Corrective steps to be taken to prevent recurrence; and
The projected date for achieving full compliance. The Authority may require responses to notices of violation to be under oath.
"NRC" means the US Nuclear Regulatory Commission or its duly authorized representatives.
"Occupational dose" means the dose received by an individual in the course of employment, education or training, in which the individual's assigned duties for the licensee or registrant involve exposure to sources of radiation, whether or not the sources of radiation are in the possession of the licensee, registrant, or other person. Occupational dose does not include doses received from background radiation, or from any medical administration the individual has received, from exposure to individuals administered radioactive material and released in accordance with U.S. Nuclear Regulatory Commission Regulations, from voluntary participation in medical research programs, or as a member of the public.
“Office of Engineering” means the office in the Delaware Division of Public Health that reviews radiation shielding plans and/or design plans and issues an Approval to Construct letter for new radiation source facilities or rooms.
“Office of Radiation Control” means the office in the Delaware Division of Public Health which carries out the Delaware Radiation Control Regulations, issues radiation source facility registration permits, and performs on-site inspections of new and existing radiation machine facilities to determine compliance.
“Owner/Leasee” means the person/individual who owns/leases the radiation source. An out-of-state owner shall authorize a manager to sign the application form.
"Package" means the packaging together with its radioactive contents as presented for transport.
"Particle accelerator" [See "Accelerator"].
"Person" means any individual, corporation, partnership, firm, association, trust, estate, public or private institution, group, agency, political subdivision of this State, any other State or political subdivision or agency thereof, and any legal successor, representative, agent, or agency of the foregoing, [but shall not include federal government agencies].
"Personnel monitoring equipment" [See "Individual monitoring devices"].
"Physician" means an allopathic doctor of medicine and surgery or a doctor of osteopathic medicine and surgery who is registered and certified to practice medicine pursuant to Title 24 Delaware Code, Chapter 17, Medical Practice Act, as amended.
“Planned special exposure” means an infrequent exposure to radiation, separate from and in addition to the annual dose limits.
“Podiatrist” means a person who is qualified to practice podiatry and is licensed under Title 24 Delaware Code, Chapter 5, Podiatry, as amended.
"Positive pressure respirator" means a respirator in which the pressure inside the respiratory inlet covering exceeds the ambient air pressure outside the respirator.
"Powered air-purifying respirator (PAPR)" means an air-purifying respirator that uses a blower to force the ambient air through air-purifying elements to the inlet covering.
"Pressure demand respirator" means a positive pressure atmosphere-supplying respirator that admits breathing air to the facepiece when the positive pressure is reduced inside the facepiece by inhalation.
“Principal Supervisor” means the State-Licensed Practitioner responsible for initiating use of x-ray equipment or other device generating ionizing radiation in the healing arts.
"Protective apron" means an apron made of radiation-attenuating materials used to reduce exposure to radiation.
"Public dose" means the dose received by a member of the public from exposure to sources of radiation released by the licensee or registrant, or to any other source of radiation under the control of the licensee or registrant. Public dose does not include occupational dose, or doses received from background radiation, from any medical administration the individual has received, from exposure to individuals administered radioactive material and released in accordance with U.S. Nuclear Regulatory Commission Regulations, or from voluntary participation in medical research programs.
“Qualified expert” means an individual who has satisfactorily fulfilled the training and experience requirements consistent with achieving a level of competency sufficient to function effectively in the position for which registration is sought. Such individuals must demonstrate to the satisfaction of the Agency their qualifications, for example, individuals certified in the appropriate field by the American Board of Radiology, or the American Board of Health Physics, or the American Board of Medical Physics, or those having equivalent qualifications. With reference to the calibration of radiation therapy equipment, an individual, in addition to the above qualifications, must be qualified in accordance with 4465 Part F and 4465 Part X of these regulations, as amended.
“Qualified Medical Physicist” means an individual qualified in accordance with Regulation 4465, Part X, Therapeutic Radiation Machines, Section 3.4, as amended. who meets each of the following credentials:
1. Has earned a master's and/or doctoral degree in physics, medical physics, biophysics, radiological physics, medical health physics, or equivalent disciplines from an accredited college or university; and
2. Has been granted certification in the specific subfield(s) of medical physics with its associated medical health physics aspects by an appropriate national certifying body and abides by the certifying body's requirements for continuing education; and/or
3. Is credentialed in accordance with Part X, subsection 3.4, as amended.
“Qualitative fit test (QLFT)" means a pass/fail fit test to assess the adequacy of respirator fit that relies on the individual’s response to the test agent.
"Quality factor" (Q) means the modifying factor, listed in Tables I and II of A.13, that is used to derive dose equivalent from absorbed dose.
"Quantitative fit test (QNFT)" means an assessment of the adequacy of respirator fit by numerically measuring the amount of leakage into the respirator.
"Rad" means the traditional unit of absorbed dose. One rad is equal to an absorbed dose of 100 erg per gram or 0.01 joule per kilogram (0.01 gray).
"Radiation" means alpha particles, beta particles, gamma rays, x rays, neutrons, high-speed electrons, high-speed protons, and other particles capable of producing ions. For purposes of these regulations, ionizing radiation is an equivalent term. Radiation, as used in these regulations, does not include non-ionizing radiation, such as radiowaves or microwaves, visible, infrared, or ultraviolet light.
"Radiation area" means any area, accessible to individuals, in which radiation levels could result in an individual receiving a dose equivalent in excess of 0.05 mSv (0.005 rem) in 1 hour at 30 centimeters from the source of radiation or from any surface that the radiation penetrates.
"Radiation dose" [See "Dose"].
"Radiation machine" means any device capable of producing ionizing radiation except those devices with radioactive material as the only source of radiation.
“Radiation Safety Officer” or RSO for a radiation machine facility means an individual assigned to perform RSO duties who has training and experience in the safe and effective use of radiation machines, their potential radiation hazards, and emergency precautions applicable to the type of activity or facility to which the RSO is assigned.
"Radiation Technician" means any individual who has not graduated from an approved program in radiation technology, but has passed an Authority approved examination.
"Radiation Technologist" means any individual who has successfully completed a JRCERT/JRCCVT approved program in radiation technology and has passed a national certification examination in his/her field of specialization.
"Radiation Technology" means the use of a radioactive substance or equipment emitting ionizing radiation on humans for diagnostic or therapeutic purposes.
"Radioactive material" means any solid, liquid or gas which emits radiation spontaneously.
"Radioactivity" means the transformation of unstable atomic nuclei by the emission of radiation.
"Radiobioassay" [See "Bioassay"].
"Registrant" means any person who is registered with the Agency and is legally obligated to register with the Agency pursuant to these regulations and the Act.
“Registration" means registration with the Agency in accordance with the regulations adopted by the Agency.
"Regulations of the Department of Transportation" means the regulations in 49 CFR Parts 100-189, as amended.
"Rem" means the traditional unit of any of the quantities expressed as dose equivalent. The dose equivalent in rem is equal to the absorbed dose in rad multiplied by the quality factor. (1 rem = 0.01 Sv)
"Research and development" means (1) theoretical analysis, exploration, or experimentation; or (2) the extension of investigative findings and theories of a scientific or technical nature into practical application for experimental and demonstration purposes, including the experimental production and testing of models, devices, equipment, materials, and processes. Research and development does not include the internal or external administration of radiation or radioactive material to human beings in the healing arts.
"Residual radioactivity" means radioactivity in structures, materials, soils, groundwater, and other media at a site resulting from activities under the licensee’s control. This includes radioactivity from all licensed and unlicensed sources used by the licensee, but excludes background radiation. It also includes radioactive materials remaining at the site as a result of routine or accidental releases of radioactive materials at the site and previous burials at the site, even if those burials were made in accordance with the provisions of Part D of these regulations.
"Restricted area" means an area, access to which is limited by the licensee or registrant for the purpose of protecting individuals against undue risks from exposure to sources of radiation. Restricted area does not include areas used as residential quarters, but separate rooms in a residential building may be set apart as a restricted area.
"Roentgen" means the traditional unit of exposure. One roentgen (R) equals 2.58E-4 coulombs per kilogram of air (see "Exposure" and Part A.9.1 of this part.)
“State Radiation Control Act” or “the Act” means Title 16 Delaware Code, Chapter 74, Radiation Control, as amended.
"Sealed source" means any encapsulated radioactive material, which has been constructed in such a manner as to prevent the escape of any radioactive material.
"Sealed Source and Device Registry (SSD)" means the national registry that contains the registration certificates, maintained by the Nuclear Regulatory Commission (NRC), that summarize the radiation safety information for sealed sources and devices, and describe the licensing and use conditions approved for the product.
"Self-contained breathing apparatus (SCBA)" means an atmosphere-supplying respirator for which the breathing air source is designed to be carried by the user.
"Shallow dose equivalent" (Hs), which applies to the external exposure of the skin or an extremity, means the dose equivalent at a tissue depth of 0.007 centimeter (7 mg/cm2) averaged over the contiguous 10 square centimeters of skin receiving the highest exposure.
"SI" means the abbreviation for Standard Internationale, the International Metric System of Measurement.
"Sievert" means the Standard Internationale (SI) unit of any of the quantities expressed as dose equivalent. The dose equivalent in sievert is equal to the absorbed dose in gray multiplied by the quality factor. (1 Sv = 100 rem)
"Source material" means:
(1) Uranium or thorium, or any combination thereof, in any physical or chemical form; or
(2) Ores that contain by weight one-twentieth of 1 percent (0.05 percent) or more of uranium, thorium or any combination of uranium and thorium. Source material does not include special nuclear material.
"Source material milling" means any activity that results in the production of byproduct material as defined by definition (2) of byproduct material, of this part.
"Source of radiation" means any radioactive material or any device or equipment emitting, or capable of producing, radiation.
"Source traceability" means the ability to show that a radioactive source has been calibrated either by the national standards laboratory of the National Institute of Standards and Technology, or by a laboratory which participates in a continuing measurement quality assurance program with National Institute of Standards and Technology or other equivalent national or international program.
"Special form radioactive material" means radioactive material that satisfies the following conditions:
(1) It is either a single solid piece or is contained in a sealed capsule that can be opened only by destroying the capsule;
(2) The piece or capsule has at least one dimension not less than 5 millimeters (0.2 inch); and
(3) It satisfies the test requirements specified by the Nuclear Regulatory Commission. A special form encapsulation designed in accordance with the Nuclear Regulatory Commission requirements in effect on June 30, 1983, and constructed prior to July 1, 1985, may continue to be used. A special form encapsulation either designed or constructed after June 30, 1985, must meet requirements of this definition applicable at the time of its design or construction.
"Special nuclear material" means:
(1) Plutonium, uranium-233, uranium enriched in the isotope 233 or in the isotope 235, and any other material that the Nuclear Regulatory Commission, pursuant to the provisions of section 51 of the Atomic Energy Act of 1954, as amended, determines to be special nuclear material, but does not include source material; or
(2) Any material artificially enriched by any of the foregoing but does not include source material.
"Special nuclear material in quantities not sufficient to form a critical mass" means uranium enriched in the isotope U-235 in quantities not exceeding 350 grams of contained U-235; uranium-233 in quantities not exceeding 200 grams; plutonium in quantities not exceeding 200 grams; or any combination of them in accordance with the following formula:
For each kind of special nuclear material, determine the ratio between the quantity of that special nuclear material and the quantity specified above for the same kind of special nuclear material.
The sum of such ratios for all of the kinds of special nuclear material in combination shall not exceed 1. For example, the following quantities in combination would not exceed the limitation and are within the formula:

“Standard Internationale (SI)” means the international metric systems of measurement.
"Supplied-air respirator (SAR)" means an atmosphere-supplying respirator for which the source of breathing air is not designed to be carried by the user.
"Survey" means an evaluation of the radiological conditions and potential hazards incident to the production, use, transfer, release, disposal, or presence of sources of radiation. When appropriate, such evaluation includes, but is not limited to, tests, physical examinations, and measurements of levels of radiation or concentrations of radioactive material present.
"Test" means the process of verifying compliance with an applicable regulation.
"These regulations" means all parts of The Delaware Radiation Control Regulations 4465, as amended.
"Tight-fitting facepiece" means a respiratory inlet covering that forms a complete seal with the face.
"Total effective dose equivalent" (TEDE) means the sum of the deep dose equivalent for external exposures and the committed effective dose equivalent for internal exposures.
"Total organ dose equivalent" (TODE) means the sum of the deep dose equivalent and the committed dose equivalent to the organ receiving the highest dose as described in subsection 39.1.6 of these regulations.
"Traceable to a National Standard" [See "Instrument traceability" or "Source traceability"].
"Unrefined and unprocessed ore" means ore in its natural form prior to any processing such as grinding, roasting, beneficiating, or refining.
"Unrestricted area" means an area, access to which is neither limited nor controlled by the licensee or registrant. For purposes of these regulations, "uncontrolled area" is an equivalent term.
"User seal check (fit check)" means an action conducted by the respirator user to determine if the respirator is properly seated to the face. Examples include negative pressure check, positive pressure check, irritant smoke check, or isoamyl acetate check.
"Very high radiation area" means an area, accessible to individuals, in which radiation levels from radiation sources external to the body could result in an individual receiving an absorbed dose in excess of 500 rads (5 grays) in 1 hour at 1 meter from a source of radiation or 1 meter from any surface that the radiation penetrates.22/
“Veterinarian” shall mean a person who has received a degree in veterinary medicine from a school of veterinary medicine, per Title 24 Delaware Code, Chapter 33, Veterinarians, as amended.
"Waste" means those low-level radioactive wastes that are acceptable for disposal in a land disposal facility. For the purposes of this definition, low-level waste has the same meaning as in the Low-Level Radioactive Waste Policy Act, P.L. 96-573, as amended by P.L. 99-240, effective January 15, 1986; that is, radioactive waste (a) not classified as high-level radioactive waste, spent nuclear fuel, or byproduct material as defined in Section 11e.(2) of the Atomic Energy Act, as amended (uranium or thorium tailings and waste) and (b) classified as low-level radioactive waste consistent with existing law and in accordance with (a) by the Nuclear Regulatory Commission.
"Waste handling licensees" mean persons licensed to receive and store radioactive wastes prior to disposal and/or persons licensed to dispose of radioactive waste.
"Week" means 7 consecutive days starting on Sunday.
"Whole body" means, for purposes of external exposure, head, trunk including male gonads, arms above the elbow, or legs above the knee.
"Worker" means an individual engaged in activities under a license or registration issued by the Agency and controlled by a licensee or registrant, including but not limited to employees, but does not include the licensee or registrant.
"Working level" (WL) means any combination of short-lived radon daughters in 1 liter of air that will result in the ultimate emission of 1.3E+5 MeV of potential alpha particle energy. The short-lived radon daughters of radon-222 are polonium-218, lead-214, bismuth-214, and polonium-214; and those of radon-220 are polonium-216, lead-212, bismuth-212, and polonium-212.
"Working level month" (WLM) means an exposure to 1 working level for 170 hours. 2,000 working hours per year divided by 12 months per year is approximately equal to 170 hours per month.
"Year" means the period of time beginning in January used to determine compliance with the provisions of these regulations. The licensee or registrant may change the starting date of the year used to determine compliance by the licensee or registrant provided that the change is made at the beginning of the year. If a licensee or registrant changes in a year, the licensee or registrant shall assure that no day is omitted or duplicated in consecutive years.
4.1 Any existing license or registration condition that is more restrictive than Part D remains in force until there is an amendment or renewal of the license or registration.
4.2 If a license or registration condition exempts a licensee or registrant from a provision of Part D in effect on or before the effective date of these regulations, it also exempts the licensee or registrant from the corresponding provision of Part D.
4.3 If a license or registration condition cites provisions of Part D in effect prior to the effective date of these regulations, which do not correspond to any provisions of Part D, the license or registration condition remains in force until there is an amendment or renewal of the license or registration that modifies or removes this condition.
Radiation Protection Programs
5.1 Each licensee or registrant shall develop, document, and implement a radiation protection program sufficient to ensure compliance with the provisions of Section 35.0 for recordkeeping requirements relating to these programs.
5.2 The licensee or registrant shall use, to the extent practical, procedures and engineering controls based upon sound radiation protection principles to achieve occupational doses and doses to members of the public that are as low as is reasonably achievable (ALARA).
5.3 The licensee or registrant shall, at intervals not to exceed 12 months, review the radiation protection program content and implementation.
5.4 To implement the ALARA requirements of subsection 5.2, and notwithstanding the requirements in Section 13.0, a constraint on air emissions of radioactive material to the environment, excluding Radon-222 and its daughters, shall be established by licensees other than those subject to 10 CFR Part 50.34a of the USNRC regulations, such that the individual member of the public likely to receive the highest dose will not be expected to receive a total effective dose equivalent in excess of 0.1 millisievert (10 mrem) per year from these emissions. If a licensee subject to this requirement exceeds this dose constraint, the licensee shall report the exceedance as provided in Section 47.0 and promptly take appropriate corrective action to ensure against recurrence.
Occupational Dose Limits
6.1 The licensee or registrant shall control the occupational dose to individual adults, except for planned special exposures pursuant to Section 10.0, to the following dose limits:
6.1.1 An annual limit, which is the more limiting of:
6.1.1.1 The total effective dose equivalent being equal to 0.05 Sievert (5 rem, or 5000 millirem); or
6.1.1.2 The sum of the deep dose equivalent and the committed dose equivalent to any individual organ or tissue other than the lens of the eye being equal to 0.5 Sievert (50 rem, or 50,000 millirem).
6.1.2 The annual limits to the lens of the eye, to the skin, and to the extremities which are:
6.1.2.1 A lens dose equivalent of 0.15 Sievert (15 rem, or 15,000 millirem); and
6.1.2.2 A shallow dose equivalent of 0.5 Sievert (50 rem, or 50,000 millirem) to the skin or to any extremity.
6.2 Doses received in excess of the annual limits, including doses received during accidents, emergencies, and planned special exposures, shall be subtracted from the limits for planned special exposures that the individual may receive during the current year and during the individual's lifetime. See subsections 10.1.5.1 and 10.1.5.2.
6.3 The assigned deep dose equivalent and shallow dose equivalent shall be for the portion of the body receiving the highest exposure:
6.3.1 The deep dose equivalent, lens dose equivalent and shallow dose equivalent may be assessed from surveys or other radiation measurements for the purpose of demonstrating compliance with the occupational dose limits, if the individual monitoring device was not in the region of highest potential exposure, or the results of individual monitoring are unavailable; or
6.3.2 When a protective apron is worn while working with medical fluoroscopic equipment and monitoring is conducted as specified in subsection 17.1.1.5, the effective dose equivalent for external radiation shall be determined as follows:
6.3.2.1 When only one individual monitoring device is used and it is located at the neck (collar) outside the protective apron, the reported deep dose equivalent shall be the effective dose equivalent for external radiation;
6.3.2.2 When only one individual monitoring device is used and it is located at the neck (collar) outside the protective apron, and the reported dose exceeds 25 percent of the limit specified in subsection 6.1 the reported deep dose equivalent value multiplied by 0.30 shall be the effective dose equivalent for external radiation; or
6.3.2.3 When individual monitoring devices are worn, both under the protective apron at the waist and outside the protective apron at the neck, the effective dose equivalent for external radiation shall be assigned the value of the sum of the deep dose equivalent reported for the individual monitoring device located at the waist under the protective apron multiplied by 1.5 and the deep dose equivalent reported for the individual monitoring device located at the neck outside the protective apron multiplied by 0.04.
6.4 Derived air concentration (DAC) and annual limit on intake (ALI) values are specified in Table I of Appendix B and may be used to determine the individual's dose and to demonstrate compliance with the occupational dose limits. See Section 39.0.
6.5 In addition to the annual dose limits, the licensee or registrant shall limit the soluble uranium intake by an individual to 10 milligrams in a week in consideration of chemical toxicity. See footnote c/ of Appendix B.
6.6 The licensee or registrant shall reduce the dose that an individual may be allowed to receive in the current year by the amount of occupational dose received while employed by any other person during the current year. See Section 37.0.
7.1 If the licensee or registrant is required to monitor pursuant to both subsections 17.1.1 and 17.1.2 the licensee or registrant shall demonstrate compliance with the dose limits by summing external and internal doses. If the licensee or registrant is required to monitor only pursuant to subsection 17.1.1 or only pursuant to subsection 17.1.2, then summation is not required to demonstrate compliance with the dose limits. The licensee or registrant may demonstrate compliance with the requirements for summation of external and internal doses pursuant to subsections 7.2, 7.3 and 7.4. The dose equivalents for the lens of the eye, the skin, and the extremities are not included in the summation, but are subject to separate limits.
7.2 Intake by Inhalation. If the only intake of radionuclides is by inhalation, the total effective dose equivalent limit is not exceeded if the sum of the deep dose equivalent divided by the total effective dose equivalent limit, and one of the following, does not exceed unity:
7.2.1 The sum of the fractions of the inhalation ALI for each radionuclide; or
7.2.2 The total number of derived air concentration-hours (DAC- hours) for all radionuclides divided by 2,000; or
7.2.3 The sum of the calculated committed effective dose equivalents to all significantly irradiated organs or tissues (T) calculated from bioassay data using appropriate biological models and expressed as a fraction of the annual limit. For purposes of this requirement, an organ or tissue is deemed to be significantly irradiated if, for that organ or tissue, the product of the weighting factors, wT, and the committed dose equivalent, HT,50, per unit intake is greater than 10 percent of the maximum weighted value of HT,50, that is, wTHT,50, per unit intake for any organ or tissue.
7.3 Intake by Oral Ingestion. If the occupationally exposed individual receives an intake of radionuclides by oral ingestion greater than 10 percent of the applicable oral ALI, the licensee or registrant shall account for this intake and include it in demonstrating compliance with the limits.
7.4 Intake through Wounds or Absorption through Skin. The licensee or registrant shall evaluate and, to the extent practical, account for intakes through wounds or skin absorption. The intake through intact skin has been included in the calculation of DAC for hydrogen-3 and does not need to be evaluated.
8.1 Licensees or registrants shall, when determining the dose from airborne radioactive material, include the contribution to the deep dose equivalent, lense dose equivalent, and shallow dose equivalent from external exposure to the radioactive cloud. See Appendix B, footnotes a/ and b/.
8.2 Airborne radioactivity measurements and DAC values shall not be used as the primary means to assess the deep dose equivalent when the airborne radioactive material includes radionuclides other than noble gases or if the cloud of airborne radioactive material is not relatively uniform. The determination of the deep dose equivalent to an individual shall be based upon measurements using instruments or individual monitoring devices.
9.1 For purposes of assessing dose used to determine compliance with occupational dose equivalent limits, the licensee or registrant shall, when required pursuant to Section 17.0, take suitable and timely measurements of:
9.1.1 Concentrations of radioactive materials in air in work areas;
9.1.2 Quantities of radionuclides in the body;
9.1.3 Quantities of radionuclides excreted from the body; or
9.1.4 Combinations of these measurements.
9.2 Unless respiratory protective equipment is used, as provided in Section 24.0, or the assessment of intake is based on bioassays, the licensee or registrant shall assume that an individual inhales radioactive material at the airborne concentration in which the individual is present.
9.3 When specific information on the physical and biochemical properties of the radionuclides taken into the body or the behavior of the material in an individual is known, the licensee or registrant may:
9.3.1 Use that information to calculate the committed effective dose equivalent, and, if used, the licensee or registrant shall document that information in the individual's record;
9.3.2 Upon prior approval of the Agency, adjust the DAC or ALI values to reflect the actual physical and chemical characteristics of airborne radioactive material, for example, aerosol size distribution or density; and
9.3.3 Separately assess the contribution of fractional intakes of Class D, W, or Y compounds of a given radionuclide to the committed effective dose equivalent. See Appendix B.
9.4 If the licensee or registrant chooses to assess intakes of Class Y material using the measurements given in 9.1.2 or 9.1.3, the licensee or registrant may delay the recording and reporting of the assessments for periods up to 7 months, unless otherwise required by Sections 46.0 or 47.0. This delay permits the licensee or registrant to make additional measurements basic to the assessments.If the identity and concentration of each radionuclide in a mixture are known, the fraction of the DAC applicable to the mixture for use in calculating DAC- hours shall be either:
9.4.1 The sum of the ratios of the concentration to the appropriate DAC value, that is, D, W, or Y, from Appendix B for each radionuclide in the mixture; or
9.4.2 The ratio of the total concentration for all radionuclides in the mixture to the most restrictive DAC value for any radionuclide in the mixture.
9.5 If the identity of each radionuclide in a mixture is known, but the concentration of one or more of the radionuclides in the mixture is not known, the DAC for the mixture shall be the most restrictive DAC of any radionuclide in the mixture.
9.6 When a mixture of radionuclides in air exists, a licensee or registrant may disregard certain radionuclides in the mixture if:
9.6.1 The licensee or registrant uses the total activity of the mixture in demonstrating compliance with the dose limits in Section 6.0 and in complying with the monitoring requirements in subsection 7.2;
9.6.2 The concentration of any radionuclide disregarded is less than 10 percent of its DAC; and
9.6.3 The sum of these percentages for all of the radionuclides disregarded in the mixture does not exceed 30 percent.
9.7 When determining the committed effective dose equivalent, the following information may be considered:
9.7.1 In order to calculate the committed effective dose equivalent, the licensee or registrant may assume that the inhalation of one ALI, or an exposure of 2,000 DAC- hours, results in a committed effective dose equivalent of 0.05 Sievert (5 rem, or 5000 millirem) for radionuclides that have their ALIs or DACs based on the committed effective dose equivalent;
9.7.2 For an ALI and the associated DAC determined by the nonstochastic organ dose limit of 0.5 Sievert (50 rem, or 50,000 millirem), the intake of radionuclides that would result in a committed effective dose equivalent of 0.05 Sievert (5 rem, or 5000 millirem), that is, the stochastic ALI, is listed in parentheses in Table I of Appendix B. The licensee or registrant may, as a simplifying assumption, use the stochastic ALI to determine committed effective dose equivalent. However, if the licensee or registrant uses the stochastic ALI, the licensee or registrant shall also demonstrate that the limit in subsection 6.1.1.2 is met.
10.1 A licensee or registrant may authorize an adult worker to receive doses in addition to and accounted for separately from the doses received under the limits specified in Section 6.0 provided that each of the following conditions is satisfied:
10.1.1 The licensee or registrant authorizes a planned special exposure only in an exceptional situation when alternatives that might avoid the dose estimated to result from the planned special exposure are unavailable or impractical;
10.1.2 The licensee or registrant, and employer if the employer is not the licensee or registrant, specifically authorizes the planned special exposure, in writing, before the exposure occurs;
10.1.3 Before a planned special exposure, the licensee or registrant ensures that each individual involved is:
10.1.3.1 Informed of the purpose of the planned operation;
10.1.3.2 Informed of the estimated doses and associated potential risks and specific radiation levels or other conditions that might be involved in performing the task; and
10.1.3.3 Instructed in the measures to be taken to keep the dose ALARA considering other risks that may be present;
10.1.4 Prior to permitting an individual to participate in a planned special exposure, the licensee or registrant ascertains prior doses as required by subsection 37.2 during the lifetime of the individual for each individual involved;
10.1.5 Subject to subsection 6.2, the licensee or registrant shall not authorize a planned special exposure that would cause an individual to receive a dose from all planned special exposures and all doses in excess of the limits to exceed:
10.1.5.1 The numerical values of any of the dose limits in subsection 6.1 in any year; and
10.1.5.2 Five times the annual dose limits in subsection 6.1 during the individual's lifetime;
10.1.6 The licensee or registrant maintains records of the conduct of a planned special exposure in accordance with Section 38.0 and submits a written report in accordance with Section 48.0;
10.1.7 The licensee or registrant records the best estimate of the dose resulting from the planned special exposure in the individual's record and informs the individual, in writing, of the dose within 30 days from the date of the planned special exposure. The dose from planned special exposures shall not be considered in controlling future occupational dose of the individual pursuant to subsection 6.1 but shall be included in evaluations required by subsections 10.1.4 and 10.1.5.
The annual occupational dose limits for minors are 10 percent of the annual occupational dose limits specified for adult workers in Section 6.0.
12.1 The licensee or registrant shall ensure that the dose equivalent to an embryo/fetus during the entire pregnancy, due to occupational exposure of a declared pregnant woman, does not exceed 5 millisievert (0.5 rem, or 500 millirem). See subsection 39.4 for recordkeeping requirements. See Appendix D for “Sample Letter for Declaring Pregnancy - Confidential, Protected Health Information”.
12.2 The licensee or registrant shall make efforts to avoid substantial variation*/ above a uniform monthly exposure rate to a declared pregnant woman so as to satisfy the limit in subsection 12.1.
12.3 The dose equivalent to the embryo/fetus is the sum of:
12.3.1 The deep dose equivalent to the declared pregnant woman; and
12.3.2 The dose equivalent resulting from radionuclides in the embryo/fetus and radionuclides in the declared pregnant woman.
12.4 If the dose equivalent to the embryo/fetus is found to have exceeded 5 millisieverts (0.5 rem, or 500 millirem), or is within 0.5 millisieverts (0.05 rem, or 50 millirem) of this dose, by the time the woman declares the pregnancy to the licensee or registrant, the licensee or registrant shall be deemed to be in compliance with subsection 12.1. if the additional dose to the embryo/fetus does not exceed 0.5 millisievert (0.05 rem, or 50 millirem) during the remainder of the pregnancy.
Radiation Dose Limits for
Individual Members of the Public
13.1 Each licensee or registrant shall conduct operations so that:
13.1.1 The total effective dose equivalent to individual members of the public from the licensed or registered operation does not exceed 1 millisievert (0.1 rem, or 100 millirem) in a year, exclusive of the dose contribution from background radiation, from any medical administration the individual has received, from exposure to individuals administered radioactive material and released in accordance with their health care providers ALARA license conditions, from voluntary participation in medical research programs, and from the licensee's or registrant's disposal of radioactive material into sanitary sewerage in accordance with D.2003;**/ and
13.1.2 The dose in any unrestricted area from external sources exclusive of the dose contributions from patients administered radioactive material and released in accordance with their health care providers ALARA license conditions, does not exceed 0.02 millisievert (0.002 rem, or 2 millirem) in any one hour; and
13.2 If the licensee or registrant permits members of the public to have access to restricted areas, the limits for members of the public continue to apply to those individuals.
13.3 A licensee, registrant, or an applicant for a license or registration may apply for prior Agency authorization to operate up to an annual dose limit for an individual member of the public of 5 millisievert (0.5 rem, or 500 millirem). This application shall include the following information:
13.3.1 Demonstration of the need for and the expected duration of operations in excess of the limit in subsection 13.1;
13.3.2 The licensee's or registrant's program to assess and control dose within the 5 millisieverts (0.5 rem or 500 millirem) annual limit; and
13.3.3 The procedures to be followed to maintain the dose as low as is reasonably achievable (ALARA).
13.4 In addition to the requirements of Part D, a licensee or registrant subject to the provisions of the Environmental Protection Agency's generally applicable environmental radiation standards in 40 CFR 190 shall comply with those standards.
13.5 The Agency may impose additional restrictions on radiation levels in unrestricted areas and on the total quantity of radionuclides that a licensee or registrant may release in effluents in order to restrict the collective dose.
14.1 The licensee or registrant shall make or cause to be made surveys of radiation levels in unrestricted areas and radioactive materials in effluents released to unrestricted areas to demonstrate compliance with the dose limits for individual members of the public in Section 13.0
14.2 A licensee or registrant shall show compliance with the annual dose limit in Section 13.0 by:
14.2.1 Demonstrating by measurement or calculation that the total effective dose equivalent to the individual likely to receive the highest dose from the licensed or registered operation does not exceed the annual dose limit; or
14.2.2 Demonstrating that:
14.2.2.1 The annual average concentrations of radioactive material released in gaseous and liquid effluents at the boundary of the unrestricted area do not exceed the values specified in Table II of Appendix B; and
14.2.2.2 If an individual were continuously present in an unrestricted area, the dose from external sources would not exceed 0.02 millisievert (0.002 rem, or 2 millirem) in an hour and 0.5 millisievert (0.05 rem, or 50 millirem) in a year.
14.3 Upon approval from the Agency, the licensee or registrant may adjust the effluent concentration values in Appendix B, Table II, for members of the public, to take into account the actual physical and chemical characteristics of the effluents, such as aerosol size distribution, solubility, density, radioactive decay equilibrium, and chemical form.
Testing for Leakage or Contamination of Sealed Sources
The licensee or registrant in possession of any sealed source shall perform leak testing of sealed sources in accordance with their radioactive material license conditions.
Surveys and Monitoring
16.1 Each licensee or registrant shall make, or cause to be made, surveys that:
16.1.1 Are necessary for the licensee or registrant to comply with Part D; and
16.1.2 Are necessary under the circumstances to evaluate:
16.1.2.1 The magnitude and extent of radiation levels;
16.1.2.2 Concentrations or quantities of radioactive material; and
16.1.2.3 The potential radiological hazards.
16.2 The licensee or registrant shall ensure that instruments and equipment used for quantitative radiation measurements, for example, dose rate and effluent monitoring, are calibrated at intervals not to exceed 12 months for the radiation measured, except when a more frequent interval is specified in another applicable Part of these regulations or a license condition.
16.3 All personnel dosimeters, except for direct and indirect reading pocket ionization chambers and those dosimeters used to measure the dose to any extremity, that require processing to determine the radiation dose and that are used by licensees and registrants to comply with Section 6.0, with other applicable provisions of these regulations, or with conditions specified in a license or registration shall be processed and evaluated by a dosimetry processor:
16.3.1 Holding current personnel dosimetry accreditation from the National Voluntary Laboratory Accreditation Program of the National Institute of Standards and Technology; and
16.3.2 Approved in this accreditation process for the type of radiation or radiations included in the National Voluntary Laboratory Accreditation Program that most closely approximates the type of radiation or radiations for which the individual wearing the dosimeter is monitored.
16.4 The licensee or registrant shall ensure that adequate precautions are taken to prevent a deceptive exposure of an individual monitoring device.
17.1 Each licensee or registrant shall monitor exposures from sources of radiation at levels sufficient to demonstrate compliance with the occupational dose limits of Part D. As a minimum:
17.1.1 Each licensee or registrant shall monitor occupational exposure to radiation from radiation sources under its control and shall supply and require the use of individual monitoring devices by:
17.1.1.1 Adults likely to receive, in 1 year from sources external to the body, a dose in excess of 10 percent of the limits in subsection 6.1;
17.1.1.2 Minors likely to receive, in 1 year from sources external to the body, a deep dose equivalent in excess of 1 millisievert (0.1 rem, or 100 millirem), a lens dose equivalent in excess of 1.5 millisievert (0.15 rem, or 150 millirem), or a shallow dose equivalent to the skin or to the extremities in excess of 5 millisievert (0.5 rem, or 500 millirem);
17.1.1.3 Declared pregnant women likely to receive during the entire pregnancy, from radiation sources external to the body, a deep dose equivalent in excess of 1 millisievert (0.1 rem, or 100 millirem);
17.1.1.4 Individuals entering a high or very high radiation area; and
17.1.1.5 Individuals working with medical fluoroscopic equipment.
17.1.1.5.1 An individual monitoring device used for the dose to an embryo/fetus of a declared pregnant woman, pursuant to subsection 12.1, shall be located under the protective apron at the waist.
17.1.1.5.2 An individual monitoring device used for lense dose equivalent shall be for external radiation pursuant to subsection 6.3.2, it shall be located at the neck (collar) outside the protective apron. When a second individual monitoring device is used for the same purpose, it shall be located under the protective apron at the waist. The second individual monitoring device is required for a declared pregnant woman.
17.1.2 Each licensee or registrant shall monitor, to determine compliance with Section 9.0, the occupational intake of radioactive material by and assess the committed effective dose equivalent to:
17.1.2.1 Adults likely to receive, in 1 year, an intake in excess of 10 percent of the applicable ALI in Table I, Columns 1 and 2, of Appendix B;
17.1.2.2 Minors likely to receive, in 1 year, a committed effective dose equivalent in excess of 0.1 millisievert (0.01 rem, or 100 millirem); and
17.1.2.3 Declared pregnant women likely to receive, during the entire pregnancy, a committed dose equivalent in excess of 1 millisievert (0.1 rem, or 100 millirem).
18.1 Each licensee or registrant shall ensure that individuals who are required to monitor occupational doses in accordance with subsection 17.1.1 wear individual monitoring devices as follows:
18.1.1 An individual monitoring device used for monitoring the dose to the whole body shall be worn at the unshielded location of the whole body likely to receive the highest exposure. When a protective apron is worn, the location of the individual monitoring device is typically at the neck (collar);
18.1.2 An individual monitoring device used for monitoring the dose to an embryo/fetus of a declared pregnant woman, pursuant to subsection 12.1, shall be located at the waist under any protective apron being worn by the woman;
18.1.3 An individual monitoring device used for monitoring the lens dose equivalent, to demonstrate compliance with subsection 6.1.2.1, shall be located at the neck (collar), outside any protective apron being worn by the monitored individual, or at an unshielded location closer to the eye;
18.1.4 An individual monitoring device used for monitoring the dose to the extremities, to demonstrate compliance with subsection 6.1.2.2, shall be worn on the extremity likely to receive the highest exposure. Each individual monitoring device shall be oriented to measure the highest dose to the extremity being monitored.
Control of Exposure from External Sources in Restricted Areas
19.1 The licensee or registrant shall ensure that each entrance or access point to a high radiation area has one or more of the following features:
19.1.1 A control device that, upon entry into the area, causes the level of radiation to be reduced below that level at which an individual might receive a deep dose equivalent of 1 millisievert (0.1 rem, or 100 millirem) in 1 hour at 30 centimeters from the source of radiation or from any surface that the radiation penetrates;
19.1.2 A control device that energizes a conspicuous visible or audible alarm signal so that the individual entering the high radiation area and the supervisor of the activity are made aware of the entry; or
19.1.3 Entryways that are locked, except during periods when access to the areas is required, with control over each individual entry.
19.2 In place of the controls required by subsection 19.1 for a high radiation area, the licensee or registrant may substitute continuous direct or electronic surveillance that is capable of preventing unauthorized entry.
19.3 The licensee or registrant may apply to the Agency for approval of alternative methods for controlling access to high radiation areas.
19.4 The licensee or registrant shall establish the controls required by subsections 19.1 and 19.3 in a way that does not prevent individuals from leaving a high radiation area.
19.5 The licensee or registrant is not required to control entrance or access to rooms or other areas in hospitals solely because of the presence of patients containing radioactive material, provided that there are personnel in attendance who are taking the necessary precautions to prevent the exposure of individuals to radiation or radioactive material in excess of the established limits in Part D and to operate within the ALARA provisions of the licensee's or registrant's radiation protection program.
19.6 The registrant is not required to control entrance or access to rooms or other areas containing sources of radiation capable of producing a high radiation area as described in Section 19.0 if the registrant has met all the specific requirements for access and control specified in other applicable Parts of these regulations, such as, Part E for industrial radiography, Part F for X-rays in the healing arts, and Part I for particle accelerators.
20.1 In addition to the requirements in Section 19.0, the licensee or registrant shall institute measures to ensure that an individual is not able to gain unauthorized or inadvertent access to areas in which radiation levels could be encountered at 5 gray (500 rad) or more in 1 hour at 1 meter from a source of radiation or any surface through which the radiation penetrates. This requirement does not apply to rooms or areas in which diagnostic X-ray systems are the only source of radiation, or to non-self-shielded irradiators.
20.2 The registrant is not required to control entrance or access to rooms or other areas containing sources of radiation capable of producing a very high radiation area as described in subsection 20.1. if the registrant has met all the specific requirements for access and control specified in other applicable Parts of these regulations, such as, Part E for industrial radiography, Part F for X-rays in the healing arts, and Part I for particle accelerators.
21.1 Section 21.0 applies to licensees or registrants with sources of radiation in non-self- shielded irradiators. Section 21.0 does not apply to sources of radiation that are used in teletherapy, in industrial radiography, or in completely self- shielded irradiators in which the source of radiation is both stored and operated within the same shielding radiation barrier and, in the designed configuration of the irradiator, is always physically inaccessible to any individual and cannot create high levels of radiation in an area that is accessible to any individual.
21.2 Each area in which there may exist radiation levels in excess of 5 gray (500 rad) in 1 hour at 1 meter from a source of radiation that is used to irradiate materials shall meet the following requirements:
21.2.1 Each entrance or access point shall be equipped with entry control devices which:
21.2.1.1 Function automatically to prevent any individual from inadvertently entering a very high radiation area;
21.2.1.2 Permit deliberate entry into the area only after a control device is actuated that causes the radiation level within the area, from the source of radiation, to be reduced below that at which it would be possible for an individual to receive a deep dose equivalent in excess of 1 millisievert (0.1 rem, or 100 millirem) in 1 hour; and
21.2.1.3 Prevent operation of the source of radiation if it would produce radiation levels in the area that could result in a deep dose equivalent to an individual in excess of 1 millisievert (0.1 rem, or 100 millirem) in 1 hour.
21.2.2 Additional control devices shall be provided so that, upon failure of the entry control devices to function as required by subsection 21.2.1:
21.2.2.1 The radiation level within the area, from the source of radiation, is reduced below that at which it would be possible for an individual to receive a deep dose equivalent in excess of 1 millisievert (0.1 rem, or 100 millirem) in 1 hour; and
21.2.2.2 Conspicuous visible and audible alarm signals are generated to make an individual attempting to enter the area aware of the hazard and at least one other authorized individual, who is physically present, familiar with the activity, and prepared to render or summon assistance, aware of the failure of the entry control devices.
21.2.3 The licensee or registrant shall provide control devices so that, upon failure or removal of physical radiation barriers other than the sealed source's shielded storage container:
21.2.3.1 The radiation level from the source of radiation is reduced below that at which it would be possible for an individual to receive a deep dose equivalent in excess of 1 millisievert (0.1 rem, or 100 millirem) in 1 hour; and
21.2.3.2 Conspicuous visible and audible alarm signals are generated to make potentially affected individuals aware of the hazard and the licensee or registrant or at least one other individual, who is familiar with the activity and prepared to render or summon assistance, aware of the failure or removal of the physical barrier.
21.2.4 When the shield for stored sealed sources is a liquid, the licensee or registrant shall provide means to monitor the integrity of the shield and to signal, automatically, loss of adequate shielding.
21.2.5 Physical radiation barriers that comprise permanent structural components, such as walls, that have no credible probability of failure or removal in ordinary circumstances need not meet the requirements of subsections 21.2.3 and 21.2.4.
21.2.6 Each area shall be equipped with devices that will automatically generate conspicuous visible and audible alarm signals to alert personnel in the area before the source of radiation can be put into operation and in time for any individual in the area to operate a clearly identified control device, which must be installed in the area and which can prevent the source of radiation from being put into operation.
21.2.7 Each area shall be controlled by use of such administrative procedures and such devices as are necessary to ensure that the area is cleared of personnel prior to each use of the source of radiation.
21.2.8 Each area shall be checked by a radiation measurement to ensure that, prior to the first individual's entry into the area after any use of the source of radiation, the radiation level from the source of radiation in the area is below that at which it would be possible for an individual to receive a deep dose equivalent in excess of 1 millisievert (0.1 rem or 100 millirem) in 1 hour.
21.2.9 The entry control devices required in subsection 21.2.1 shall be tested for proper functioning. See Section 42.0 for recordkeeping requirements.
21.2.9.1 Testing shall be conducted prior to initial operation with the source of radiation on any day, unless operations were continued uninterrupted from the previous day;
21.2.9.2 Testing shall be conducted prior to resumption of operation of the source of radiation after any unintentional interruption; and
21.2.9.3 The licensee or registrant shall submit and adhere to a schedule for periodic tests of the entry control and warning systems.
21.2.10 The licensee or registrant shall not conduct operations, other than those necessary to place the source of radiation in safe condition or to effect repairs on controls, unless control devices are functioning properly.
21.2.11 Entry and exit portals that are used in transporting materials to and from the irradiation area, and that are not intended for use by individuals, shall be controlled by such devices and administrative procedures as are necessary to physically protect and warn against inadvertent entry by any individual through these portals. Exit portals for irradiated materials shall be equipped to detect and signal the presence of any loose radioactive material that is carried toward such an exit and automatically to prevent loose radioactive material from being carried out of the area.
21.3 Licensees, registrants, or applicants for licenses or registrations for sources of radiation within the purview of subsection 21.2 which will be used in a variety of positions or in locations, such as open fields or forests, that make it impracticable to comply with certain requirements of subsection 21.2, such as those for the automatic control of radiation levels, may apply to the Agency for approval of alternative safety measures. Alternative safety measures shall provide personnel protection at least equivalent to those specified in subsection 21.2. At least one of the alternative measures shall include an entry-preventing interlock control based on a measurement of the radiation that ensures the absence of high radiation levels before an individual can gain access to the area where such sources of radiation are used.
21.4 The entry control devices required by subsections 21.2 and 21.3 shall be established in such a way that no individual will be prevented from leaving the area.
Respiratory Protection and Controls to Restrict
Internal Exposure in Restricted Areas
The licensee or registrant shall use, to the extent practical, process or other engineering controls, such as, containment, decontamination or ventilation, to control the concentrations of radioactive material in air.
23.1 When it is not practicable to apply process or other engineering controls to control the concentrations of radioactive material in air to values below those that define an airborne radioactivity area, the licensee or registrant shall, consistent with maintaining the total effective dose equivalent ALARA, increase monitoring and limit intakes by one or more of the following means:
23.1.1 Control of access;
23.1.2 Limitation of exposure times;
23.1.3 Use of respiratory protection equipment; or
23.1.4 Other controls.
23.2 If the licensee performs an ALARA analysis to determine whether or not respirators should be used, the licensee may also consider the impact of respirator use on workers' industrial health and safety.
24.1 If the licensee or registrant uses respiratory protection equipment to limit intakes pursuant to Section 23.0:
24.1.1 Except as provided in subsection 24.1.2, the licensee or registrant shall use only respiratory protection equipment that is tested and certified by the National Institute for Occupational Safety and Health;
24.1.2 If the licensee or registrant wishes to use equipment that has not been tested or certified by the National Institute for Occupational Safety and Health, or for which there is no schedule for testing or certification, the licensee shall submit an application to the Agency for authorized use of this equipment, except as otherwise noted in this Part. The application must include evidence that the material and performance characteristics of the equipment are capable of providing the proposed degree of protection under anticipated conditions of use. This must be demonstrated either by the licensee's or registrant's testing or on the basis of reliable test information;
24.1.3 The licensee or registrant shall implement and maintain a respiratory protection program that includes:
24.1.3.1 Air sampling sufficient to identify the potential hazard, permit proper equipment selection, and estimate doses;
24.1.3.2 Surveys and bioassays, as necessary, to evaluate actual intakes;
24.1.3.3 Testing of respirators for operability (user seal check for face sealing devices and functional check for others) immediately prior to each use; and
24.1.3.4 Written procedures regarding:
24.1.3.4.1 Monitoring, including air sampling and bioassays;
24.1.3.4.2 Supervision and training or respirator users;
24.1.3.4.3 Fit testing;
24.1.3.4.4 Respirator selection;
24.1.3.4.5 Breathing air quality;
24.1.3.4.6 Inventory and control;
24.1.3.4.7 Storage, issuance, maintenance, repair, testing, and quality assurance of respiratory protection equipment;
24.1.3.4.8 Recordkeeping; and
24.1.3.4.9 Limitations on periods of respirator use and relief from respirator use.
24.1.3.5 Determination by a physician that the individual user is medically fit to use the respiratory protection equipment before:
24.1.3.5.1 The initial fitting of a face sealing respirator;
24.1.3.5.2 Before the first field use of non- face sealing respirators, and
24.1.3.5.3 Either every 12 months thereafter, or periodically at a frequency determined by a physician.
24.1.3.6 Fit testing, with a fit factor 10 times the APF for negative pressure devices, and a fit factor 500 for any positive pressure, continuous flow, and pressure-demand devices, before the first field use of tight fitting, face sealing respirators and periodically thereafter at a frequency not to exceed 1 year. Fit testing must be performed with the facepiece operating in the negative pressure mode.
24.1.4 The licensee or registrant shall advise each respirator user that the user may leave the area at any time for relief from respirator use in the event of equipment malfunction, physical or psychological distress, procedural or communication failure, significant deterioration of operating conditions, or any other conditions that might require such relief.
24.1.5 The licensee or registrant shall also consider limitations appropriate to the type and mode of use. When selecting respiratory devices the licensee or registrant shall provide for vision correction, adequate communication, low temperature work environments and the concurrent use of other safety or radiological protection equipment. The licensee or registrant shall use equipment in such a way as not to interfere with the proper operation of the respirator.
24.1.6 Standby rescue persons are required whenever one-piece atmosphere-supplying suits, or any combination of supplied air respiratory protection device and personnel protective equipment are used from which an unaided individual would have difficulty extricating himself or herself. The standby persons must be equipped with respiratory protection devices or other apparatus appropriate for the potential hazards. The standby rescue persons shall observe or otherwise maintain continuous communication with the workers (visual, voice, signal line, telephone, radio, or other suitable means), and be immediately available to assist them in case of a failure of the air supply or for any other reason that requires relief from distress. A sufficient number of standby rescue persons must be immediately available to assist all users of this type of equipment and to provide effective emergency rescue if needed.
24.1.7 Atmosphere-supplying respirators must be supplied with respirable air of grade D quality or better as defined by the Compressed Gas Association in publication G-7.1, "Commodity Specification for Air," 1997 and included in the regulations of the Occupational Safety and Health Administration (29 CFR 1910.134(i)(1)(ii)(A) through (E). Grade D quality air criteria include:
24.1.7.1 Oxygen content (v/v) of 19.5-23.5%;
24.1.7.2 Hydrocarbon (condensed) content of 5 milligrams per cubic meter of air or less;
24.1.7.3 Carbon Monoxide (CO) content of 10 ppm or less;
24.1.7.4 Carbon Dioxide content of 1,000 ppm or less; and
24.1.7.5 Lack of noticeable odor.
24.1.8 The licensee shall ensure that no objects, materials or substances, such as facial hair, or any conditions that interfere with the face- facepiece seal or valve function, and that are under the control of the wearer, are present between the skin of the wearer’s face and the sealing surface of a tight- fitting respirator facepiece.
24.1.9 In estimating the dose to individuals from intake of airborne radioactive materials, the concentration of radioactive material in the air that is inhaled when respirators are worn is initially assumed to be the ambient concentration in air without the respiratory protection, divided by the assigned protection factor. If the dose is later found to be greater than the estimated dose, the corrected value must be used. If the dose is later found to be less than the estimated dose, the corrected value may be used.
25.1 The Agency may impose restrictions in addition to the provisions of Sections 23.0 and 24.0, and Appendix A of this Part, in order to:
25.1.1 Ensure that the respiratory protection program of the licensee is adequate to limit doses to individuals from intakes of radioactive materials consistent with maintaining total effective dose equivalent ALARA; and
25.1.2 Limit the extent to which a licensee may use respiratory protection equipment instead of process or other engineering controls.
26.1 The licensee or registrant shall obtain authorization from the Agency before using assigned respiratory protection factors in excess of those specified in Appendix A. The Agency may authorize a licensee or registrant to use higher protection factors on receipt of an application that:
26.1.1 Describes the situation for which a need exists for higher protection factors; and
26.1.2 Demonstrates that the respiratory protection equipment provides these higher protection factors under the proposed conditions of use.
Storage and Control of Licensed or Registered
Sources of Radiation
27.1 The licensee or registrant shall secure licensed or registered radioactive material from unauthorized removal or access.
27.2 The licensee or registrant shall maintain constant surveillance, and use engineering controls or devices, or administrative procedures to prevent unauthorized use of licensed or registered radioactive material that is in an unrestricted area and that is not in storage.
27.3 The registrant shall secure registered radiation machines from unauthorized removal.
27.4 The registrant shall use devices or administrative procedures to prevent unauthorized use of registered radiation machines.
Precautionary Procedures
28.1 Standard Radiation Symbol. Unless otherwise authorized by the Agency, the symbol prescribed by this section shall use the colors magenta, or purple, or black on yellow background. The symbol prescribed is the three-bladed design as follows:
Figure 1. Radiation Symbol.
1. Cross-hatched area is to be magenta, or purple, or black, and
2. The background is to be yellow.
28.2 Exception to Color Requirements for Standard Radiation Symbol. Notwithstanding the requirements of subsection 28.1, licensees or registrants are authorized to label sources, source holders, or device components containing sources of radiation that are subjected to high temperatures, with conspicuously etched or stamped radiation caution symbols and without a color requirement.
28.3 Additional Information on Signs and Labels. In addition to the contents of signs and labels prescribed in Part D, the licensee or registrant may provide, on or near the required signs and labels, additional information, as appropriate, to make individuals aware of potential radiation exposures and to minimize the exposures.
29.1 Posting of Radiation Areas. The licensee or registrant shall post each radiation area with a conspicuous sign or signs bearing the radiation symbol and the words "CAUTION, RADIATION AREA."
29.2 Posting of High Radiation Areas. The licensee or registrant shall post each high radiation area with a conspicuous sign or signs bearing the radiation symbol and the words "CAUTION, HIGH RADIATION AREA" or "DANGER, HIGH RADIATION AREA."
29.3 Posting of Very High Radiation Areas. The licensee or registrant shall post each very high radiation area with a conspicuous sign or signs bearing the radiation symbol and words " DANGER, VERY HIGH RADIATION AREA."
29.4 Posting of Airborne Radioactivity Areas. The licensee or registrant shall post each airborne radioactivity area with a conspicuous sign or signs bearing the radiation symbol and the words "CAUTION, AIRBORNE RADIOACTIVITY AREA" or "DANGER, AIRBORNE RADIOACTIVITY AREA."
29.5 Posting of Areas or Rooms in which Licensed or Registered Material is Used or Stored. The licensee or registrant shall post each area or room in which there is used or stored an amount of licensed or registered material exceeding 10 times the quantity of such material specified in Appendix C with a conspicuous sign or signs bearing the radiation symbol and the words "CAUTION, RADIOACTIVE MATERIAL(S)" or "DANGER, RADIOACTIVE MATERIAL(S)."
30.1 A licensee or registrant is not required to post caution signs in areas or rooms containing sources of radiation for periods of less than 8 hours, if each of the following conditions is met:
30.1.1 The sources of radiation are constantly attended during these periods by an individual who takes the precautions necessary to prevent the exposure of individuals to sources of radiation in excess of the limits established in Part D; and
30.1.2 The area or room is subject to the licensee’s or registrant’s control.
30.2 A room or area is not required to be posted with a caution sign because of the presence of a sealed source provided the radiation level at 30 centimeters from the surface of the sealed source container or housing does not exceed 0.05 millisievert (0.005 rem or 5 millirem) per hour.
30.3 A room or area is not required to be posted with a caution sign because of the presence of radiation machines used solely for diagnosis in the healing arts.
30.4 Rooms in hospitals or clinics that are used for teletherapy are exempt from the requirement to post caution signs under Section 29.0 if:
30.4.1 Access to the room is controlled pursuant to the providers ALARA license conditions; and
30.4.2 Personnel in attendance take necessary precautions to prevent the inadvertent exposure of workers, other patients, and members of the public to radiation in excess of the limits established in this Part.
Each registrant shall ensure that each radiation machine is labeled in a conspicuous manner which cautions individuals that radiation is produced when it is energized.
32.1 A licensee or registrant is not required to label radiation machines if:
32.1.1 The area or room is subject to the licensee’s or registrant’s control and precautions are taken to prevent the exposure of individuals to sources of radiation in excess of the limits established in Part D.
Waste Disposal
Compliance with Environmental and Health Protection Regulations. Nothing in these regulations relieves the licensee or registrant from complying with other applicable Federal, State and local regulations governing any other toxic or hazardous properties of materials disposed of by the licensee or registrant.
Records
34.1 Each licensee or registrant shall use the Standard Internationale (SI) units becquerel, gray, sievert and coulomb per kilogram, or the special units curie, rad, rem and roentgen, including multiples and subdivisions, and shall clearly indicate the units of all quantities on records required by Part D.
34.2 The licensee or registrant shall make a clear distinction among the quantities entered on the records required by Part D, such as, total effective dose equivalent shallow dose equivalent, lens dose equivalent, deep dose equivalent, or committed effective dose equivalent.
35.1 Each licensee or registrant shall maintain records of the radiation protection program, including:
35.1.1 The provisions of the program; and
35.1.2 Audits and other reviews of program content and implementation.
35.2 The licensee or registrant shall retain the records required by subsection 35.1.1 until the Agency terminates each pertinent license or registration requiring the record. The licensee or registrant shall retain the records required by subsection 35.1.2 for 3 years after the record is made.
36.1 Each licensee or registrant shall maintain records showing the results of surveys and calibrations required by Section 16.0. The licensee or registrant shall retain these records for 3 years after the record is made.
36.2 The licensee or registrant shall retain each of the following records until the Agency terminates each pertinent license or registration requiring the record:
36.2.1 Records of the results of surveys to determine the dose from external sources of radiation used, in the absence of or in combination with individual monitoring data, in the assessment of individual dose equivalents;
36.2.2 Records of the results of measurements and calculations used to determine individual intakes of radioactive material and used in the assessment of internal dose;
36.2.3 Records showing the results of air sampling, surveys, and bioassays required pursuant to subsections 24.1.3.1 and 24.1.3.2; and
36.2.4 Records of the results of measurements and calculations used to evaluate the release of radioactive effluents to the environment.
37.1 For each individual who is likely to receive, in a year, an occupational dose requiring monitoring pursuant to Section 17.0, the licensee or registrant shall:
37.1.1 Determine the occupational radiation dose received during the current year; and
37.1.2 Attempt to obtain the records of cumulative occupational radiation dose.
37.2 Prior to permitting an individual to participate in a planned special exposure, the licensee or registrant shall determine:
37.2.1 The internal and external doses from all previous planned special exposures; and
37.2.2 All doses in excess of the limits, including doses received during accidents and emergencies, received during the lifetime of the individual; and
37.3 In complying with the requirements of subsection 37.1, a licensee or registrant may:
37.3.1 Accept, as a record of the occupational dose that the individual received during the current year, a written signed statement from the individual, or from the individual's most recent employer for work involving radiation exposure, that discloses the nature and the amount of any occupational dose that the individual received during the current year; and
37.3.2 Accept, as the record of cumulative radiation dose, an up-to-date Agency Form Y (Part J, Appendix B) or equivalent, signed by the individual and countersigned by an appropriate official of the most recent employer for work involving radiation exposure, or the individual's current employer, if the individual is not employed by the licensee or registrant; and.
37.3.3 Obtain reports of the individual's dose equivalent(s) from the most recent employer for work involving radiation exposure, or the individual's current employer, if the individual is not employed by the licensee or registrant, by telephone, email, facsimile, other electronic media or letter. The licensee or registrant shall request a written verification of the dose data if the authenticity of the transmitted report cannot be established.
37.4 The licensee or registrant shall record the exposure history, as required by subsection 37.1, on Agency Form Y, (Part J, Appendix B) or other clear and legible record, of all the information required on that form.
37.4.1 The form or record shall show each period in which the individual received occupational exposure to radiation or radioactive material and shall be signed by the individual who received the exposure. For each period for which the licensee or registrant obtains reports, the licensee or registrant shall use the dose shown in the report in preparing Agency form Y (Part J, Appendix B) or equivalent. For any period in which the licensee or registrant does not obtain a report, the licensee or registrant shall place a notation on Agency Form Y or equivalent indicating the periods of time for which data are not available.
37.4.2 For the purposes of complying with this requirement, licensees or registrants are not required to partition historical dose between external dose equivalent(s) and internal committed dose equivalent(s). Further, occupational exposure histories obtained and recorded on Agency Form Y (Part J, Appendix B) or equivalent before July 10, 2002, would not have included effective dose equivalent, but may be used in the absence of specific information on the intake of radionuclides by the individual.
37.5 If the licensee or registrant is unable to obtain a complete record of an individual's current and previously accumulated occupational dose, the licensee or registrant shall assume:
37.5.1 In establishing administrative controls pursuant to subsection 6.6 for the current year, that the allowable dose limit for the individual is reduced by 12.5 millisievert (1.25 rem, or 1250 millirem) for each quarter for which records were unavailable and the individual was engaged in activities that could have resulted in occupational radiation exposure; and
37.5.2 That the individual is not available for planned special exposures.
37.6 The licensee or registrant shall retain the records on Agency Form Y (Part J, Appendix B) or equivalent until the Agency terminates each pertinent license or registration requiring this record. The licensee or registrant shall retain records used in preparing Agency Form Y (Part J, Appendix B) or equivalent for 3 years after the record is made.
37.7 Upon termination of the license or registration, the licensee or registrant shall permanently store records on Agency Form Y (Part J, Appendix B) or equivalent, or shall make provision with the Agency for transfer to the Agency.
38.1 For each use of the provisions of Section 10.0 for planned special exposures, the licensee or registrant shall maintain records that describe:
38.1.1 The exceptional circumstances requiring the use of a planned special exposure;
38.1.2 The name of the management official who authorized the planned special exposure and a copy of the signed authorization;
38.1.3 What actions were necessary;
38.1.4 Why the actions were necessary;
38.1.5 What precautions were taken to assure that doses were maintained ALARA;
38.1.6 What individual and collective doses were expected to result; and
38.1.7 The doses actually received in the planned special exposure.
38.2 The licensee or registrant shall retain the records until the Agency terminates each pertinent license or registration requiring these records.
38.3 Upon termination of the license or registration, the licensee or registrant shall permanently store records on Agency Form Y (Part J, Appendix B) or equivalent, or shall make provision with the Agency for transfer to the Agency.
39.1 Recordkeeping Requirement. Each licensee or registrant shall maintain records of doses received by all individuals for whom monitoring was required pursuant to Section 17.0, and records of doses received during planned special exposures, accidents, and emergency conditions. Assessments of dose equivalent and records made using units in effect before need not be changed. These records shall include, when applicable:
39.1.1 The deep dose equivalent to the whole body, lens dose equivalent, shallow dose equivalent to the skin, and shallow dose equivalent to the extremities;
39.1.2 The estimated intake of radionuclides, see Section 7.0;
39.1.3 The committed effective dose equivalent assigned to the intake of radionuclides;
39.1.4 The specific information used to calculate the committed effective dose equivalent pursuant to subsections 9.1 and 9.3 and when required by Section 17.0;
39.1.5 The total effective dose equivalent when required by Section 7.0; and
39.1.6 The total of the deep dose equivalent and the committed dose to the organ receiving the highest total dose.
39.2 Recordkeeping Frequency. The licensee or registrant shall make entries of the records specified in subsection 39.1 at intervals not to exceed 1 year.
39.3 Recordkeeping Format. The licensee or registrant shall maintain the records specified in subsection 39.1 on Agency Form Z (Part J, Appendix C), in accordance with the instructions for Agency Form Z (Part J, Appendix C), or in clear and legible records containing all the information required by Agency Form Z (Part J, Appendix C).
39.4 The licensee or registrant shall maintain the records of dose to an embryo/fetus with the records of dose to the declared pregnant woman. The declaration of pregnancy, including the estimated date of conception, shall also be kept on file, but must be maintained as confidential records, accessible only to the registrant or licensee Radiation Safety Officer, the declared pregnant woman, and medical personnel authorized to access her confidential health records.
39.5 The licensee or registrant shall retain each required form or record until the Agency terminates each pertinent license or registration requiring the record.
39.6 Upon termination of the license or registration, the licensee or registrant shall permanently store records on Agency Form Y (Part J, Appendix B) or equivalent, or shall make provision with the Agency for transfer to the Agency.
40.1 Each licensee or registrant shall maintain records sufficient to demonstrate compliance with the dose limit for individual members of the public. See Section 13.0.
40.2 The licensee or registrant shall retain the records required by subsection 40.1 until the Agency terminates each pertinent license or registration requiring the record.
Compliance with Environmental and Health Protection Regulations. Nothing in these regulations relieves the licensee or registrant from complying with other applicable Federal, State and local regulations governing any other toxic or hazardous properties of materials that may be disposed of by the licensee or registrant.
42.1 Each licensee or registrant shall maintain records of tests made pursuant to subsection 21.2.9 on entry control devices for very high radiation areas. These records must include the date, time, and results of each such test of function.
42.2 The licensee or registrant shall retain the records required by subsection 42.1 for 3 years after the record is made.
Each record required by Part D shall be legible throughout the specified retention period. The record shall be the original or a reproduced copy or a microform, provided that the copy or microform is authenticated by authorized personnel and that the microform is capable of producing a clear copy throughout the required retention period or the record may also be stored in electronic media with the capability for producing legible, accurate, and complete records during the required retention period. Records, such as letters, drawings, and specifications, shall include all pertinent information, such as stamps, initials, and signatures. The licensee shall maintain adequate safeguards against tampering with and loss of records.
Records of tests for leakage or contamination of sealed sources required by Part D, Section 15.0 shall be kept on file in accordance with the facility radioactive material license.
Reports
45.1 Telephone Reports. Each licensee or registrant shall report to the Agency by telephone immediately after its occurrence becomes known to the registrant, any stolen, lost, or missing radiation machine.
45.2 Written Reports. Each licensee or registrant required to make a report pursuant to subsection 45.1 shall, within 30 days after making the telephone report, make a written report to the Agency setting forth the following information:
45.2.1 A description of the licensed or registered source of radiation involved, including, for radiation machines, the manufacturer, model and serial number, type and maximum energy of radiation emitted;
45.2.2 A description of the circumstances under which the loss or theft occurred;
45.2.3 A statement of disposition, or probable disposition, of the registered source of radiation involved;
45.2.4 Exposures of individuals to radiation, and circumstances under which the exposures occurred;
45.2.5 Actions that have been taken, or will be taken, to recover the source of radiation; and
45.2.6 Procedures or measures that have been, or will be, adopted to ensure against a recurrence of the loss or theft of licensed or registered sources of radiation.
45.3 Subsequent to filing the written report, the licensee or registrant shall also report additional substantive information on the loss or theft within 30 days after the licensee or registrant learns of such information.
45.4 The licensee or registrant shall prepare any report filed with the Agency pursuant to this section so that names of individuals who may have received exposure to radiation are stated in a separate and detachable portion of the report.
46.1 Immediate Notification. Notwithstanding other requirements for notification, each licensee or registrant shall immediately report each event involving a source of radiation possessed by the licensee or registrant that may have caused or threatens to cause any of the following conditions:
46.1.1 An individual to receive:
46.1.1.1 A total effective dose equivalent of 0.25 sievert (25 rem, or 25,000 millirem) or more;
46.1.1.2 A lense dose equivalent of 0.75 sievert (75 rem, or 75,000 millirem) or more; or
46.1.1.3 A shallow dose equivalent to the skin or extremities or a total organ dose equivalent of 2.5 gray (250 rad) or more; or
46.1.2 The release of radioactive material, inside or outside of a restricted area, so that, had an individual been present for 24 hours, the individual could have received an intake five times the occupational ALI. This provision does not apply to locations where personnel are not normally stationed during routine operations, such as hot-cells or process enclosures.
46.2 Twenty-Four Hour Notification. Each licensee or registrant shall, within 24 hours of discovery of the event, report to the Agency each event involving loss of control of a licensed or registered source of radiation possessed by the licensee or registrant that may have caused, or threatens to cause, any of the following conditions:
46.2.1 An individual to receive, in a period of 24 hours:
46.2.1.1 A total effective dose equivalent exceeding 0.05 sievert (5 rem, or 5000 millirem);
46.2.1.2 A lense dose equivalent exceeding 0.15 sievert (15 rem, or 15,000 millirem);
46.2.1.3 A shallow dose equivalent to the skin or extremities or a total organ dose equivalent exceeding 0.5 sievert (50 rem, or 50,000 millirem); or
46.2.2 The release of radioactive material, inside or outside of a restricted area, so that, had an individual been present for 24 hours, the individual could have received an intake in excess of one occupational ALI. This provision does not apply to locations where personnel are not normally stationed during routine operations, such as hot-cells or process enclosures.
46.3 Licensees or registrants shall make the reports required by subsections 46.1 and 46.2 by initial contact by telephone to the Agency and shall confirm the initial contact immediately by email, express mail or facsimile to the Agency. The Agency shall reply to the written notification within 24 hours to discuss timing of follow-up action by the Agency.
46.4 The licensee or registrant shall prepare each report filed with the Agency pursuant to Section 46.0 so that names of individuals who have received exposure to sources of radiation are stated in a separate and detachable portion of the report.
46.5 The provisions of Section 46.0 do not apply to doses that result from planned special exposures, provided such doses are within the limits for planned special exposures and are reported pursuant to Section 48.0.
47.1 Reportable Events. In addition to the notification required by Section 46.0, each licensee or registrant shall submit a written report within 30 days after learning of any of the following occurrences:
47.1.1 Incidents for which notification is required by Section 46.0; or
47.1.2 Doses in excess of any of the following:
47.1.2.1 The occupational dose limits for adults in Section 6.0;
47.1.2.2 The occupational dose limits for a minor in Section 11.0;
47.1.2.3 The limits for an embryo/fetus of a declared pregnant woman in Section 12.0;
47.1.2.4 The limits for an individual member of the public in Section 13.0;
47.1.2.5 Any applicable limit in the license or registration; or
47.1.2.6 The ALARA constraints for air emissions established under subsection 5.4
47.1.3 Level of radiation or concentrations of radioactive material in:
47.1.3.1 A restricted area in excess of applicable limits in the license or registration; or
47.1.3.2 An unrestricted area in excess of 10 times the applicable limit set forth in Part D or in the license or registration, whether or not involving exposure of any individual in excess of the limits in Section 13.0; or
47.1.4 For licensees subject to the provisions of the Environmental Protection Agency's generally applicable environmental radiation standards in 40 CFR 190, levels of radiation or releases of radioactive material in excess of those standards, or of license conditions related to those standards.
47.2 Contents of Reports.
47.2.1 Each report required by subsection 47.1 shall describe the extent of exposure of individuals to radiation and radioactive material, including, as appropriate:
47.2.1.1 Estimates of each individual's dose;
47.2.1.2 The levels of radiation and concentrations of radioactive material involved;
47.2.1.3 The cause of the elevated exposures, dose rates, or concentrations; and
47.2.1.4 Corrective steps taken or planned to ensure against a recurrence, including the schedule for achieving conformance with applicable limits, ALARA constraints generally applicable environmental standards, and associated license or registration conditions.
47.2.2 Each report filed pursuant to subsection 47.1 shall include for each occupationally overexposeda/ individual: the name, unique identification number such as employee or Social Security number, and date of birth. With respect to the limit for the embryo/fetus in Section 12.0, the identifiers should be those of the declared pregnant woman. The report shall be prepared so that this information is stated in a separate and detachable portion of the report.
47.3 All licensees or registrants who make reports pursuant to subsection 47.1 shall submit the report in writing to the Agency.
The licensee or registrant shall submit a written report to the Agency within 30 days following any planned special exposure conducted in accordance with Section 10.0, informing the Agency that a planned special exposure was conducted and indicating the date the planned special exposure occurred and the information required by Part D, Section 38.0.
When a licensee or registrant is required, pursuant to Part D, Sections 47.0, 48.0, or D.2206 22.6 to report to the Agency any exposure of an identified occupationally exposed individual, or an identified member of the public, to radiation or radioactive material, the licensee shall also provide a copy of the report submitted to the Agency to the individual. This report must be transmitted at a time no later than the transmittal to the Agency.
50.1 Requirements for notification and reports to individuals of exposure to radiation or radioactive material are specified in Part J, Section 4.0 of these regulations.
50.2 When a licensee or registrant is required pursuant to Section 47.0 to report to the Agency any exposure of an individual to radiation or radioactive material, the licensee or registrant shall also notify the individual. Such notice shall be transmitted at a time not later than the transmittal to the Agency, and shall comply with the provisions of Part J, Subsection 4.1 of these regulations.
Additional Requirements
Each specific licensee or registrant in possession of a radiation source shall, no less than 30 days before vacating or relinquishing possession or control of premises, notify the Agency in writing of intent to vacate. When deemed necessary by the Agency, the licensee shall decontaminate the premises in such a manner as the Agency may specify.
A PDF version of Appendices A-F for Part D is available here:
This Part establishes requirements, for which a registrant is responsible, for use of diagnostic x-ray equipment by, or under the supervision of, an individual authorized by and licensed in accordance with State statutes to engage in the healing arts or veterinary medicine. The provisions of this Part are in addition to, and not in substitution for, other applicable provisions of Parts A, B, D, J and K of the regulations. Some registrants may also be subject to the requirements of Parts I and X of the regulations.
As used in this Part, the following definitions apply:
"Accessible surface" means the external surface of the enclosure or housing of the radiation producing machine as provided by the manufacturer.
"Accessory component" means:
(1) A component used with diagnostic x-ray systems, such as a cradle or film changer, that is not necessary for the compliance of the system with applicable provisions of this Part but which requires an initial determination of compatibility with the system; or
(2) A component necessary for compliance of the system with applicable provisions of this Part but which may be interchanged with similar compatible components without affecting the system’s compliance, such as one of a set of interchangeable beam-limiting devices; or
(3) A component compatible with all x-ray systems with which it may be used and that does not require compatibility or installation instructions, such as a tabletop cassette holder.
"Air kerma" means kerma in air (see definition of Kerma).
"Air kerma rate (AKR)" means the air kerma per unit time.
"Aluminum equivalent" means the thickness of type 1100 aluminum alloy (The nominal chemical composition of type 1100 aluminum is 99.00 percent minimum aluminum, 0.12 percent copper.) affording the same attenuation, under specified conditions, as the material in question.
"Articulated joint" means a joint between two separate sections of a tabletop which joint provides the capacity of one of the sections to pivot on the line segment along which the sections join.
"Assembler" means any person engaged in the business of assembling, replacing, or installing one or more components into an x-ray system or subsystem. The term includes the owner of an x-ray system or his or her employee or agent who assembles components into an x-ray system that is subsequently used to provide professional or commercial services.
"Attenuation block" means a block or stack, having dimensions 20 centimeters by 20 centimeters by 3.8 centimeters, of type 1100 aluminum alloy (The nominal chemical composition of type 1100 aluminum is 99.00 percent minimum aluminum, 0.12 percent copper.) or other materials having equivalent attenuation.
"Automatic exposure control (AEC)" means a device which automatically controls one or more technique factors in order to obtain at a preselected location(s) a required quantity of radiation (Includes devices such as phototimers and ion chambers).
"Automatic exposure rate control (AERC)" means a device which automatically controls one or more technique factors in order to obtain, at a preselected location(s), a required quantity of radiation per unit time.
"Barrier" (See "Protective barrier").
"Beam axis" means a line from the source through the centers of the x-ray fields.
"Beam-limiting device" means a device which provides a means to restrict the dimensions of the x- ray field.
"Bone densitometry system" means a medical device which uses electronically-produced ionizing radiation to determine the density of bone structures of human patients.
"C-arm fluoroscope" means a fluoroscopic x-ray system in which the image receptor and the x-ray tube housing assembly are connected or coordinated to maintain a spatial relationship. Such a system allows a change in the direction of the beam axis with respect to the patient without moving the patient.
"Cantilevered tabletop" means a tabletop designed such that the unsupported portion can be extended at least 100 cm beyond the support.
"Cassette holder" means a device, other than a spot-film device, that supports and/or fixes the position of an x-ray film [imaging] cassette during an x-ray exposure.
"Coefficient of variation (C)" means the ratio of the standard deviation to the mean value of a population of observations. It is estimated using the following equation:
where:
 = Estimated standard deviation of the population.
= Estimated standard deviation of the population.
 = Mean value of observations in sample;
= Mean value of observations in sample;
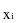 = ith observation in sample;
= ith observation in sample;
n = Number of observations sampled.
"Computed tomography (CT)" means the production of a tomogram by the acquisition and computer processing of x-ray transmission data.
"Control panel" means that part of the x-ray control upon which are mounted the switches, knobs, pushbuttons, and other hardware necessary for manually setting the technique factors.
"Cooling curve" means the graphical relationship between heat units stored and cooling time.
"Cradle" means:
(1) A removable device which supports and may restrain a patient above an x-ray table; or
(2) A device;
(i) Whose patient support structure is interposed between the patient and the image receptor during normal use;
(ii) Which is equipped with means for patient restraint; and
(iii) Which is capable of rotation about its long (longitudinal) axis.
"CT" (See "Computed tomography").
"CT gantry" means tube housing assemblies, beam-limiting devices, detectors, and the supporting structures, frames, and covers which hold and/or enclose these components.
"Cumulative air kerma" means the total air kerma accrued from the beginning of an examination or procedure and includes all contributions from fluoroscopic and radiographic irradiation.
"Detector" (See "Radiation detector")
"Diagnostic source assembly" means the tube housing assembly with a beam-limiting device attached.
"Diagnostic x-ray system" means an x-ray system designed for irradiation of any part of the human [or animal] body for the purpose of diagnosis or visualization.
"Direct scattered radiation" means that scattered radiation which has been deviated in direction only by materials irradiated by the useful beam (See "Scattered radiation").
"Dose" means the absorbed dose as defined by the International Commission on Radiation Units and Measurements. The absorbed dose, D, is the quotient of de by dm, where de is the mean energy imparted to matter of mass dm; thus D=de/dm, in units of J/kg, where the special name of the unit of absorbed dose is gray (Gy).
"Equipment" (See "X-ray equipment").
"Exposure (X)" means the quotient of dQ by dm where dQ is the absolute value of the total charge of the ions of one sign produced in air when all the electrons and positrons liberated or created by photons in air of mass dm are completely stopped in air; thus X=dQ/dm, in units of C/kg. A second meaning of exposure is the process or condition during which the x-ray tube produces x-ray radiation.
"Field emission equipment" means equipment which uses an x-ray tube in which electron emission from the cathode is due solely to the action of an electric field.
"Filter" means material placed in the useful beam to preferentially absorb selected radiations.
"Fluoroscopic air kerma display devices" means separate devices, subsystems, or components that provide the display of AKR and cumulative air kerma, respectively, required by 5.0. They include radiation detectors, if any, electronic and computer components, associated software, and data displays.
"Fluoroscopic imaging assembly" means a subsystem in which x-ray photons produce a set of fluoroscopic images or radiographic images recorded from the fluoroscopic image receptor. It includes the image receptor(s), electrical interlocks, if any, and structural material providing linkage between the image receptor and diagnostic source assembly.
"Fluoroscopic irradiation time" means the cumulative duration during an examination or procedure of operator-applied continuous pressure to the device, enabling x-ray tube activation in any fluoroscopic mode of operation.
"Fluoroscopy" means a technique for generating x-ray images and presenting them simultaneously and continuously as visible images. This term has the same meaning as the term “radioscopy” in the standards of the International Electrotechnical Commission.
"Focal spot (actual)" means the area projected on the anode of the x-ray tube bombarded by the electrons accelerated from the cathode and from which the useful beam originates.
"General purpose radiographic x-ray system" means any radiographic x-ray system which, by design, is not limited to radiographic examination of specific anatomical regions.
"Gonad shield" means a protective barrier for the testes or ovaries.
"Half-value layer (HVL)" means the thickness of specified material which attenuates the beam of radiation to an extent such that the AKR is reduced by one-half of its original value. In this definition, the contribution of all scattered radiation, other than any which might be present initially in the beam concerned, is deemed to be excluded.
"Hand-held x-ray equipment" means x-ray equipment that is designed to be hand-held during operation.
"Healing arts screening" means the testing of human beings using x-ray machines for the detection or evaluation of health indications when such tests are not specifically and individually ordered by a licensed practitioner of the healing arts legally authorized to prescribe such x-ray tests for the purpose of diagnosis or treatment.
"Heat unit" means a unit of energy equal to the product of the peak kilovoltage, milliamperes, and seconds, i.e., kVp x mA x second.
"HVL" (See "Half-value layer").
"Image intensifier" means a device, installed in its housing, which instantaneously converts an x-ray pattern into a corresponding light image of higher intensity.
"Image receptor" means any device, such as a fluorescent screen, radiographic film, x-ray image intensifier tube, solid-state detector, or gaseous detector which transforms incident x-ray photons either into a visible image or into another form which can be made into a visible image by further transformations. In those cases where means are provided to preselect a portion of the image receptor, the term “image receptor” shall mean the preselected portion of the device.
"Image receptor support device" means, for mammography x-ray systems, that part of the system designed to support the image receptor during a mammographic examination and to provide a primary protective barrier.
"Irradiation" means the exposure of matter to ionizing radiation.
"Isocenter" means the center of the smallest sphere through which the beam axis passes when the equipment moves through a full range of rotations about its common center.
"Kerma" means the quantity defined by the International Commission on Radiation Units and Measurements. The kerma, K, is the quotient of dEtr by dm, where dEtr is the sum of the initial kinetic energies of all the charged participles liberated by uncharged particles in a mass dm of material; thus K=dEtr/dm, in units of J/kg, where the special name for the unit of kerma is gray (Gy). When the material is air, the quantity is referred to as “air kerma.”
"Kilovolts peak" (See "Peak tube potential").
"kV" means kilovolts.
"kVp" (See "Peak tube potential").
"kWs" means kilowatt second.
"Last image hold (LIH) radiograph" means an image obtained either by retaining one or more fluoroscopic images, which may be temporarily integrated, at the end of a fluoroscopic exposure or by initiating a separate and distinct radiographic exposure automatically and immediately in conjunction with termination of the fluoroscopic exposure.
"Lateral fluoroscope" means the x-ray tube and image receptor combination in a biplane system dedicated to the lateral projection. It consists of the lateral x-ray tube housing assembly and the lateral image receptor that are fixed in position relative to the table with the x-ray beam axis parallel to the plane of the table.
"Lead equivalent" means the thickness of lead affording the same attenuation, under specified conditions, as the material in question.
"Leakage radiation" means radiation emanating from the diagnostic source assembly except for:
(1) The useful beam; and
(2) Radiation produced when the exposure switch or timer is not activated.
"Leakage technique factors" means the technique factors associated with the diagnostic source assembly which are used in measuring leakage radiation. They are defined as follows:
(1) For diagnostic source assemblies intended for capacitor energy storage equipment, the maximum-rated peak tube potential and the maximum-rated number of exposures in an hour for operation at the maximum-rated peak tube potential with the quantity of charge per exposure being 10 millicoulombs (or 10 mAs) or the minimum obtainable from the unit, whichever is larger;
(2) For diagnostic source assemblies intended for field emission equipment rated for pulsed operation, the maximum-rated peak tube potential and the maximum-rated number of x-ray pulses in an hour for operation at the maximum-rated peak tube potential;
(3) For all other diagnostic source assemblies, the maximum-rated peak tube potential and the maximum-rated continuous tube current for the maximum-rated peak tube potential.
"Licensed Practitioner" means an individual licensed to practice medicine, dentistry, dental hygiene, podiatry, chiropractic, or osteopathy in this State.
"Light field" means that area of the intersection of the light beam from the beam-limiting device and one of the set of planes parallel to and including the plane of the image receptor, whose perimeter is the locus of points at which the illumination is one-fourth of the maximum in the intersection.
"Line-voltage regulation" means the difference between the no-load and the load line potentials expressed as a percent of the load line potential , as follows:
Percent line-voltage regulation = 100 (Vn-Vl)/Vl
where:
Vn = No-load line potential; and
Vl = Load line potential.
"mA" means milliampere.
“mAs" means milliampere second.
"Mobile x-ray equipment" (See "X-ray equipment").
"Mode of operation" means, for fluoroscopic systems, a distinct method of fluoroscopy or radiography provided by the manufacturer and selected with a set of several technique factors or other control settings uniquely associated with the mode. The set of distinct technique factors and control settings for the mode may be selected by the operation of a single control. Examples of distinct modes of operation include normal fluoroscopy (analog or digital), high-level control fluoroscopy, cineradiography (analog and digital), digital subtraction angiography, electronic radiography using the fluoroscopic image receptor, and photospot recording. In a specific mode of operation, certain system variables affecting kerma, AKR, or image quality, such as image magnification, x-ray field size, pulse rate, pulse duration, number of pulses, source-image receptor distance (SID), or optical aperture, may be adjustable or may vary; their variation per se does not comprise a mode of operation different from the one that has been selected.
"Movable tabletop" means a tabletop which, when assembled for use, is capable of movement with respect to its supporting structure within the plane of the tabletop.
"Non-image-intensified fluoroscopy" means fluoroscopy using only a fluorescent screen.
"Patient" means an individual or animal subjected to healing arts examination, diagnosis or treatment.
"PBL" See "Positive beam limitation."
"Peak tube potential" means the maximum value of the potential difference across the x-ray tube during an exposure.
"Phantom" means a volume of material behaving in a manner similar to tissue with respect to the attenuation and scattering of radiation. This requires that both the atomic number (Z) and the density of the material be similar to that of tissue.
"PID" (See "Position indicating device").
"Portable x-ray equipment" (See "X-ray equipment").
"Position indicating device" means a device on dental x-ray equipment used to indicate the beam position and to establish a definite source-surface (skin) distance. It may or may not incorporate or serve as a beam-limiting device.
"Positive beam limitation" means the automatic or semi-automatic adjustment of an x-ray beam to the size of the selected image receptor, whereby exposures cannot be made without such adjustment.
"Primary protective barrier" means the material, excluding filters, placed in the useful beam to reduce the radiation exposure [beyond the patient and cassette holder] for protection purposes.
"Protective apron" means an apron made of radiation absorbing materials used to reduce radiation exposure.
"Protective glove" means a glove made of radiation absorbing materials used to reduce radiation exposure.
"Pulsed mode" means operation of the x-ray system such that the x-ray tube current is pulsed by the x-ray control to produce one or more exposure intervals of duration less than one-half second.
“Qualified expert” means an individual who has demonstrated to the satisfaction of the Agency that such individual possesses the knowledge and training to measure ionizing radiation, to evaluate safety techniques, and to advise regarding radiation protection needs, for example, individuals certified in the appropriate field by the American Board of Radiology, or the American Board of Health Physics, or the American Board of Medical Physics, or those having equivalent qualifications. With reference to the calibration of radiation therapy equipment, an individual, in addition to the above qualifications, must be qualified in accordance with Part X.
"Qualified medical physicist" means an individual who meets the requirements specified in Part X.
"Quick change x-ray tube" means an x-ray tube designed for use in its associated tube housing such that:
(1) The tube cannot be inserted in its housing in a manner that would result in noncompliance of the system with the requirements of 6.0;
(2) The focal spot position will not cause noncompliance with the provisions of this section or;
(3) The shielding within the tube housing cannot be displaced; and
(4) Any removal and subsequent replacement of a beam-limiting device during reloading of the tube in the tube housing will not result in noncompliance of the x-ray system with the applicable field limitation and alignment requirements of 6.0.
"Radiation detector" means a device which in the presence of radiation provides a signal or other indication suitable for use in measuring one or more quantities of incident radiation.
"Radiation therapy simulation system" means a radiographic or fluoroscopic x‑ray system intended for localizing the volume to be exposed during radiation therapy and confirming the position and size of the therapeutic irradiation field.
"Radiograph" means an image receptor on which the image is created directly or indirectly by an x-ray pattern and results in a permanent record.
"Radiography" means a technique for generating and recording an x-ray pattern for the purpose of providing the user with an image(s) after termination of the exposure.
"Rated line voltage" means the range of potentials, in volts, of the supply line specified by the manufacturer at which the x-ray machine is designed to operate.
"Rated output current" means the maximum allowable load current of the x-ray high-voltage generator.
"Rating" means the operating limits specified by the manufacturer.
"Recording" means producing a retrievable form of an image resulting from x-ray photons.
"Scan" means the complete process of collecting x-ray transmission data for the production of a tomogram. Data may be collected simultaneously during a single scan for the production of one or more tomograms.
"Scan time" means the period of time between the beginning and end of x-ray transmission data accumulation for a single scan.
"Scattered radiation" means radiation that, during passage through matter, has been deviated in direction (See "Direct scattered radiation").
"Shutter" means a device attached to the tube housing assembly which can intercept the entire cross sectional area of the useful beam and which has a lead equivalency not less than that of the tube housing assembly.
"SID" (See "Source-image receptor distance").
"Solid state x-ray imaging device" means an assembly, typically in a rectangular panel configuration, that intercepts x-ray photons and converts the photon energy into a modulated electronic signal representative of the x-ray intensity over the area of the imaging device. The electronic signal is then used to create an image for display and/or storage.
"Source" means the focal spot of the x-ray tube.
"Source-image receptor distance" means the distance from the source to the center of the input surface of the image receptor.
"Source-skin distance (SSD)" means the distance from the source to the center of the entrant x-ray field in the plane tangent to the patient skin surface.
"Spot film" means a radiograph which is made during a fluoroscopic examination to permanently record conditions which exist during that fluoroscopic procedure.
"Spot-film device" means a device intended to transport and/or position a radiographic image receptor between the x-ray source and fluoroscopic image receptor. It includes a device intended to hold a cassette over the input end of the fluoroscopic image receptor for the purpose of producing a radiograph.
"Stationary tabletop" means a tabletop which, when assembled for use, is incapable of movement with respect to its supporting structure within the plane of the tabletop.
"Stationary x-ray equipment" (See "X-ray equipment").
"Stray radiation" means the sum of leakage and scattered radiation.
"Technique factors" means the following conditions of operation:
(1) For capacitor energy storage equipment, peak tube potential in kilovolts (kV) and quantity of charge in milliampere-seconds (mAs);
(2) For field emission equipment rated for pulsed operation, peak tube potential in kV, and number of x-ray pulses;
(3) For CT equipment designed for pulsed operation, peak tube potential in kV, scan time in seconds, and either tube current in milliamperes (mA), x-ray pulse width in seconds, and the number of x-ray pulses per scan, or the product of tube current, x-ray pulse width, and the number of x-ray pulses in mAs;
(4) For CT equipment not designed for pulsed operation, peak tube potential in kV, and either tube current in mA and scan time in seconds, or the product of tube current and exposure time in mAs and the scan time when the scan time and exposure time are equivalent; and
(5) For all other equipment, peak tube potential in kV, and either tube current in mA and exposure time in seconds, or the product of tube current and exposure time in mAs.
"Tomogram" means the depiction of the x-ray attenuation properties of a section through the body.
"Tube" means an x-ray tube, unless otherwise specified.
"Tube housing assembly" means the tube housing with tube installed. It includes high-voltage and/or filament transformers and other appropriate elements when such are contained within the tube housing.
"Tube rating chart" means the set of curves which specify the rated limits of operation of the tube in terms of the technique factors.
"Useful beam" means the radiation which passes through the tube housing port and the aperture of the beam limiting device when the exposure switch or timer is activated.
"Variable-aperture beam-limiting device" means a beam-limiting device which has capacity for stepless adjustment of the x-ray field size at a given SID.
"Visible area" means that portion of the input surface of the image receptor over which incident x-ray photons are producing a visible image.
"X-ray control" means a device which controls input power to the x-ray high-voltage generator and/or the x-ray tube. It includes equipment such as timers, phototimers, automatic brightness stabilizers, and similar devices, which control the technique factors of an x-ray exposure.
"X-ray exposure control" means a device, switch, button or other similar means by which an operator initiates and/or terminates the radiation exposure. The x-ray exposure control may include such associated equipment as timers and back-up timers.
"X-ray equipment" means an x-ray system, subsystem, or component thereof. Types of x-ray equipment are as follows:
(1) "Mobile x-ray equipment" means x-ray equipment mounted on a permanent base with wheels and/or casters for moving while completely assembled.
(2) "Portable x-ray equipment" means x-ray equipment designed to be hand-carried.
(3) "Stationary x-ray equipment" means x-ray equipment which is installed in a fixed location.
"X-ray field" means that area of the intersection of the useful beam and any one of a set of planes parallel to and including the plane of the image receptor, whose perimeter is the locus of points at which the AKR is one-fourth of the maximum in the intersection.
"X-ray high-voltage generator" means a device which transforms electrical energy from the potential supplied by the x-ray control to the tube operating potential. The device may also include means for transforming alternating current to direct current, filament transformers for the x-ray tube(s), high-voltage switches, electrical protective devices, and other appropriate elements.
"X-ray subsystem" means any combination of two or more components of an x-ray system for which there are requirements specified in this section and 4.0, 5.0, 6.0.
"X-ray system" means an assemblage of components for the controlled production of x-rays. It includes minimally an x-ray high-voltage generator, an x-ray control, a tube housing assembly, a beam-limiting device, and the necessary supporting structures. Additional components which function with the system are considered integral parts of the system.
"X-ray table" means a patient support device with its patient support structure (tabletop) interposed between the patient and the image receptor during radiography and/or fluoroscopy. This includes, but is not limited to, any stretcher equipped with a radiolucent panel and any table equipped with a cassette tray (or bucky), cassette tunnel, fluoroscopic image receptor, or spot-film device beneath the tabletop.
"X-ray tube" means any electron tube which is designed for the conversion of electrical energy into x-ray energy.
3.1 Radiation Safety Requirements. The registrant, licensee, shall be responsible for directing the operation of the x-ray system(s) under his administrative control. The registrant, licensee, or the registrant's, licensee’s, agent shall assure that the requirements of these regulations are met in the operation of the x-ray system(s).
3.1.1 An x-ray system which does not meet the provisions of these regulations shall not be operated for diagnostic purposes.
3.1.2 Individuals who will be operating the x-ray systems shall meet the Agency’s qualifications to conduct the practice of radiologic technology.
3.1.3 A chart shall be provided in the vicinity of the diagnostic x-ray system's control panel which specifies, for all examinations performed with that system, the following information:
3.1.3.1 Patient's body part and anatomical size, or body part thickness, or age (for pediatrics), versus technique factors to be utilized;
3.1.3.2 Type and size of the image receptor to be used;
3.1.3.3 Type and size of the image receptor combination to be used, if any;
3.1.3.4 Source to image receptor distance to be used (except for dental intraoral radiography);
3.1.3.5 Type and location of placement of patient shielding (e.g., gonad, etc.) to be used; and
3.1.3.6 For mammography, indication of kVp/target/filter combination.
3.1.4 The registrant [licensee] of a facility shall create and make available to x-ray operators written safety procedures, including patient holding and any restrictions of the operating technique required for the safe operation of the particular x-ray system. The operator shall be able to demonstrate familiarity with these procedures.
3.1.5 Except for patients who cannot be moved out of the room, only the staff, ancillary personnel or other persons required for the medical procedure or training shall be in the room during the radiographic exposure. Other than the patient being examined:
3.1.5.1 All individuals shall be positioned such that no part of the body will be struck by the useful beam unless protected by not less than 0.5 millimeter lead equivalent material;
3.1.5.2 The x-ray operator, other staff, ancillary personnel, and other persons required for the medical procedure shall be protected from the direct scatter radiation by protective aprons or whole body protective barriers of not less than 0.25 millimeter lead equivalent material;
3.1.5.3 Human patients who cannot be removed from the room shall be protected from the direct scatter radiation by whole body protective barriers of not less than 0.25 millimeter lead equivalent material or shall be so positioned that the nearest portion of the body is at least 2 meters from both the tube head and the nearest edge of the image receptor.
3.1.6 Gonad shielding of not less than 0.5 millimeter lead equivalent material shall be used for human patients, who have not passed the reproductive age, during radiographic procedures in which the gonads are in the useful beam, except for cases in which this would interfere with the diagnostic procedure.
3.1.7 Individuals shall not be exposed to the useful beam except for healing arts purposes and unless such exposure has been authorized by a licensed practitioner of the healing arts. This provision specifically prohibits deliberate exposure for the following purposes:
3.1.7.1 Exposure of an individual for training, demonstration, or other non-healing arts purposes; and
3.1.7.2 Exposure of an individual for the purpose of healing arts screening except as authorized by 3.1.11.
3.1.8 When a patient or image receptor must be provided with auxiliary support during a radiation exposure:
3.1.8.1 Mechanical holding devices shall be used when the technique permits. The written safety procedures, required by 3.1.4, shall list individual projections where holding devices cannot be utilized;
3.1.8.2 Written safety procedures, as required by 3.1.4, shall indicate the requirements for selecting a holder and the procedure the holder shall follow;
3.1.8.3 The human holder shall be instructed in personal radiation safety and protected as required by 3.1.5.;
3.1.8.4 No individual shall be used routinely to hold image receptor or patients;
3.1.8.5 In those cases where the patient must hold the image receptor, except during intraoral examinations, any portion of the body other than the area of clinical interest struck by the useful beam shall be protected by not less than 0.5 millimeter lead equivalent material; and
3.1.8.6 Each facility shall have leaded aprons and gloves available in sufficient numbers to provide protection for all personnel who are involved with x-ray operations and who are otherwise not shielded.
3.1.9 Procedures and auxiliary equipment designed to minimize patient and personnel exposure commensurate with the needed diagnostic information shall be utilized.
3.1.9.1 The fastest imaging system consistent with the diagnostic objective of the examinations shall be used. Film cassettes without intensifying screens shall not be used for any routine diagnostic radiological imaging, with the exception of veterinary radiography and standard film packets for intraoral use in dental radiography.
3.1.9.2 The radiation exposure to the patient shall be the minimum exposure required to produce images of good diagnostic quality.
3.1.9.3 Portable or mobile x-ray equipment shall be used only for examinations where it is impractical to transfer the patient(s) to a stationary x-ray installation. The use of hand held devices must be approved by the Agency utilizing the criteria in Appendix B.
3.1.9.4 X-ray systems subject to 6.0 shall not be utilized in procedures where the source to patient distance is less than 30 centimeters, except for veterinary systems.
3.1.9.5 If grids are used between the patient and the image receptor to decrease scatter to the film and improve contrast, the grid shall:
3.1.9.5.1 Be positioned properly, i.e., tube side facing the right direction, and grid centered to the central ray;
3.1.9.5.2 If the grid is of the focused type, be of the proper focal distance for the SIDs being used.
3.1.10 All individuals who are associated with the operation of an x-ray system are subject to the requirements of D.1201, D.1207 and D.1208 of these regulations.
3.1.11 Healing Arts Screening. Any person proposing to conduct a healing arts screening program shall not initiate such a program without prior approval of the Agency. When requesting such approval, that person shall submit the information outlined in Appendix A of this Part. If any information submitted to the Agency becomes invalid or outdated, the Agency shall be immediately notified.
3.1.12 Information and Maintenance Record and Associated Information. The registrant [licensee] shall maintain the following information for each x-ray system for inspection by the Agency:
3.1.12.1 Model and serial numbers of all major components, and user's manuals for those components;
3.1.12.2 Tube rating charts and cooling curves;
3.1.12.3 Records of surveys, calibrations, maintenance, and modifications performed on the x-ray system(s); and
3.1.12.4 A copy of all correspondence with this Agency regarding that x-ray system.
3.1.13 X-Ray Utilization Record. Except for veterinary facilities, each facility shall maintain a record containing the patient's name, the type of examinations, and the dates the examinations were performed. When the patient or film must be provided with human auxiliary support, the name of the human holder should be recorded.
3.2 X-Ray Film Processing Facilities and Practices.
3.2.1 Each installation using a radiographic x-ray system and using analog image receptors (e.g. radiographic film) shall have available suitable equipment for handling and processing radiographic film in accordance with the following provisions:
3.2.1.1 Manually developed film:
3.2.1.2 Processing tanks shall be constructed of mechanically rigid, corrosion resistant material; and
3.2.1.3 The temperature of solutions in the tanks shall be maintained within the range of 60o F to 80o F (16o C to 27o C). Film shall be developed in accordance with the time-temperature relationships recommended by the film manufacturer, or, in the absence of such recommendations, with the following time-temperature chart:
Time-Temperature Chart | ||
Thermometer Reading (Degrees) | Minimum Developing Time (Minutes) | |
oC | oF | |
26.7 | 80 | 2 |
26.1 | 79 | 2 |
25.6 | 78 | 2½ |
25.0 | 77 | 2½ |
24.4 | 76 | 3 |
23.9 | 75 | 3 |
23.3 | 74 | 3½ |
22.8 | 73 | 3½ |
22.2 | 72 | 4 |
21.7 | 71 | 4 |
21.1 | 70 | 4½ |
20.6 | 69 | 4½ |
20.0 | 68 | 5 |
19.4 | 67 | 5½ |
18.9 | 66 | 5½ |
18.3 | 65 | 6 |
17.8 | 64 | 6½ |
17.2 | 63 | 7 |
16.7 | 62 | 8 |
16.1 | 61 | 8½ |
15.6 | 60 | ½ |
3.2.1.4 Devices shall be utilized which will indicate the actual temperature of the developer and signal the passage of a preset time appropriate to the developing time required.
3.2.1.5 Automatic processors and other closed processing systems: Films shall be developed in accordance with the time-temperature relationships recommended by the film manufacturer; in the absence of such recommendations, the film shall be developed using the following chart:
Developer Temperature | Minimum Immersion Timea/ | |
oC | oF | Seconds |
35.5 | 96 | 19 |
35 | 95 | 20 |
34.5 | 94 | 21 |
34 | 93 | 22 |
33.5 | 92 | 23 |
33 | 91 | 24 |
32 | 90 | 25 |
31.5 | 89 | 26 |
31 | 88 | 27 |
30.5 | 87 | 28 |
30 | 86 | 29 |
29.5 | 85 | 30 |
a/ Immersion time only, no crossover time included. | ||
3.2.1.6 Processing deviations from the requirements of 3.2.1 shall be documented by the registrant [licensee] in such manner that the requirements are shown to be met or exceeded (e.g., extended processing, and special rapid chemistry).
3.3 Other Requirements.
3.3.1 Pass boxes, if provided, shall be so constructed as to exclude light from the darkroom when cassettes are placed in or removed from the boxes, and shall incorporate adequate shielding from stray radiation to prevent exposure of undeveloped film.
3.3.2 The darkroom shall be light tight and use proper safelighting such that any film type in use exposed in a cassette to x-radiation sufficient to produce an optical density from 1 to 2 when processed shall not suffer an increase in density greater than 0.1 (0.05 for mammography) when exposed in the darkroom for 2 minutes with all safelights on. If used, daylight film handling boxes shall preclude fogging of the film.
3.3.3 Darkrooms typically used by more than one individual shall be provided a method to prevent accidental entry while undeveloped films are being handled or processed.
3.3.4 Film shall be stored in a cool, dry place and shall be protected from exposure to stray radiation. Film in open packages shall be stored in a light tight container.
3.3.5 Film cassettes and intensifying screens shall be inspected periodically and shall be cleaned and replaced as necessary to best assure radiographs of good diagnostic quality.
3.3.6 Outdated x-ray film shall not be used for diagnostic radiographs, unless the film has been stored in accordance with the manufacturer's recommendations and a sample of the film passes a sensitometric test for normal ranges of base plus fog and speed.
3.3.7 Film developing solutions shall be prepared in accordance with the directions given by the manufacturer, and shall be maintained in strength by replenishment or renewal so that full development is accomplished within the time specified by the manufacturer.
In addition to other requirements of this Part, all diagnostic x‑ray systems shall meet the following requirements:
4.1 Warning Label. The control panel containing the main power switch shall bear the warning statement, legible and accessible to view: "WARNING: This x-ray unit may be dangerous to patient and operator unless safe exposure factors, operating instructions and maintenance schedules are observed."
4.2 Leakage Radiation from the Diagnostic Source Assembly. The leakage radiation from the diagnostic source assembly measured at a distance of 1 meter in any direction from the source shall not exceed 0.88 milligray (mGy) air kerma (vice 100 milliroentgen (mR) exposure) in 1 hour when the x-ray tube is operated at its leakage technique factors. If the maximum rated peak tube potential of the tube housing assembly is greater than the maximum rated peak tube potential for the diagnostic source assembly, positive means shall be provided to limit the maximum x-ray tube potential to that of the diagnostic source assembly. Compliance shall be determined by measurements averaged over an area of 100 square centimeters with no linear dimension greater than 20 centimeters.
4.3 Radiation from Components Other Than the Diagnostic Source Assembly. The radiation emitted by a component other than the diagnostic source assembly shall not exceed an air kerma of 18 microgray (vice 2 milliroentgens exposure) in 1 hour at 5 centimeters from any accessible surface of the component when it is operated in an assembled x-ray system under any conditions for which it was designed. Compliance shall be determined by measurements averaged over an area of 100 square centimeters with no linear dimension greater than 20 centimeters.
4.4 Beam Quality.
4.4.1 Half-Value Layer (HVL).
4.4.1.1 The HVL of the useful beam for a given x-ray tube potential shall not be less than the values shown in Table 1 in this section under the heading “Specified Dental Systems,” for any dental x-ray system designed for use with intraoral image receptors and manufactured after December 1, 1980; under the heading, “I-Other X-Ray Systems,” for any dental x-ray system designed for use with intraoral image receptors and manufactured before or on December 1, 1980, and all other x-ray systems subject to this section and manufactured before June 10, 2006; and under the heading, “II-Other X-Ray Systems,” for all x-ray systems, except dental x-ray systems designed for use with intraoral image receptors, subject to this section and manufactured on or after June 10, 2006. If it is necessary to determine such half-value layer at an x-ray tube potential which is not listed in Table 1 of this section, linear interpolation or extrapolation may be made. Positive means shall be provided to ensure that at least the minimum filtration needed to achieve beam quality requirements is in the useful beam during each exposure. In the case of a system, which is to be operated with more than one thickness of filtration, this requirement can be met by a filter interlocked with the kilovoltage selector which will prevent x-ray emissions if the minimum required filtration is not in place.
X-Ray Tube Voltage (kilovolt peak) | ||||
Design Operating Range | Measured Operating Potential | Minimum HVL (mm in Aluminum) | ||
Specified Dental Systems \1\ | Other X-Ray Systems\2\ | Other X-Ray Systems\3\ | ||
Below 51 | 30 | 1.5 | 0.3 | 0.3 |
40 | 1.5 | 0.4 | 0.4 | |
50 | 1.5 | 0.5 | 0.5 | |
51 to 70 | 51 | 1.5 | 1.2 | 1.3 |
60 | 1.5 | 1.3 | 1.5 | |
70 | 1.5 | 1.5 | 1.8 | |
Above 70 | 71 | 2.1 | 2.1 | 2.5 |
80 | 2.3 | 2.3 | 2.9 | |
90 | 2.5 | 2.5 | 3.2 | |
100 | 2.7 | 2.7 | 3.6 | |
110 | 3.0 | 3.0 | 3.9 | |
120 | 3.2 | 3.2 | 4.3 | |
130 | 3.5 | 3.5 | 4.7 | |
140 | 3.8 | 3.8 | 5.0 | |
150 | 4.1 | 4.1 | 5.4 | |
\1\ Dental x-ray systems designed for use with intraoral image receptors and manufactured after December 1, 1980. \2\ Dental x-ray systems designed for use with intraoral image receptors and manufactured before or on December 1, 1980, and all other x-ray systems subject to this section and manufactured before June 10, 2006. \3\ All x-ray systems, except dental x-ray systems designed for use with intraoral image receptors, subject to this section and manufactured on or after June 10, 2006. | ||||
4.4.1.2 Optional filtration. Fluoroscopic systems manufactured on or after June 10, 2006, incorporating an x-ray tube(s) with a continuous output of 1 kilowatt or more and an anode heat storage capacity of 1 million heat units or more shall provide the option of adding x-ray filtration to the diagnostic source assembly in addition to the amount needed to meet the half-value layer provisions of this subsection. The selection of this additional x-ray filtration shall be either at the option of the user or automatic as part of the selected mode of operation. A means of indicating which combination of additional filtration is in the x-ray beam shall be provided.
4.4.1.3 Measuring compliance. For capacitor energy storage equipment, compliance shall be determined with the maximum selectable quantity of charge per exposure.
4.4.1.4 Aluminum equivalent of material between patient and image receptor. Except when used in a CT x-ray system, the aluminum equivalent of each of the items listed in Table 2 in this paragraph, which are used between the patient and the image receptor, may not exceed the indicated limits. Compliance shall be determined by x-ray measurements made at a potential of 100 kilovolts peak and with an x-ray beam that has an HVL specified in Table 1 of this section for the potential. This requirement applies to front panel(s) of cassette holders and film changers provided by the manufacturer for patient support or for prevention of foreign object intrusions. It does not apply to screens and their associated mechanical support panels or grids. Table 2 follows:
Item | Maximum Aluminum Equivalent (millimeters) |
Front panel(s) of cassette holders (total of all) Film panel(s) of film changer (total of all) Cradle Tabletop, stationary, without articulated joints Tabletop, movable, without articulated joint(s) (including stationary subtop) Tabletop, with radiolucent panel having one articulated joint Tabletop, with radiolucent panel having two or more articulated joints Tabletop, cantilevered Tabletop, radiation therapy simulator | 1.2 1.2 2.3 1.2 1.7 1.7 2.3 2.3 5.0 |
4.5 Battery charge indicator. On battery-powered generators, visual means shall be provided on the control panel to indicate whether the battery is in a state of charge adequate for proper operation.
4.6 Modification of certified diagnostic x-ray components and systems.
4.7 Diagnostic x-ray components and systems certified in accordance with 21 CFR Part 1020 shall not be modified such that the component or system fails to comply with any applicable provision of this Part.
4.8 The owner of a diagnostic x-ray system who uses the system in a professional or commercial capacity may modify the system provided the modification does not result in the failure of the system or component to comply with the applicable requirements of this Part. The owner who causes such modification need not submit the reports required by this Part, provided the owner records the date and the details of the modification in the system records and maintains this information, and provided the modification of the x-ray system does not result in a failure to comply with this Part. Such system modifications should be per suggested manufacturer’s specifications.
4.9 Multiple Tubes. Where two or more radiographic tubes are controlled by one exposure switch, the tube or tubes which have been selected shall be clearly indicated prior to initiation of the exposure. This indication shall be both on the x-ray control panel and at or near the tube housing assembly which has been selected.
4.10 Mechanical Support of Tube Head. The tube housing assembly supports shall be adjusted such that the tube housing assembly will remain stable during an exposure unless tube housing movement is a designed function of the x-ray system.
4.11 Technique Indicators.
4.11.1 For x-ray equipment capable of displaying technique factors, the technique factors to be used during an exposure shall be indicated before the exposure begins. If automatic exposure controls are used, the technique factors which are set prior to the exposure shall be indicated.
4.11.2 The requirement of 4.11.1 may be met by permanent markings on equipment having fixed technique factors. Indication of technique factors shall be visible from the operator's position except in the case of spot films made by the fluoroscopist.
4.12 Maintaining Compliance. Diagnostic x-ray systems and their associated components used on humans and certified pursuant to the Federal X-Ray Equipment Performance Standard (21 CFR Part 1020) shall be maintained in compliance with applicable requirements of that standard.
4.13 Locks. All position locking, holding, and centering devices on x-ray system components and systems shall function as intended.
The provisions of this Part apply to equipment for fluoroscopic imaging or for recording images from the fluoroscopic image receptor, except computed tomography x-ray systems manufactured on or after November 29, 1984.
5.1 Primary Protective Barrier.
5.1.1 Limitation of useful beam. The fluoroscopic imaging assembly shall be provided with a primary protective barrier which intercepts the entire cross section of the useful beam at any SID. The x-ray tube used for fluoroscopy shall not produce x-rays unless the barrier is in position to intercept the entire useful beam. The AKR due to transmission through the barrier with the attenuation block in the useful beam combined with radiation from the fluoroscopic imaging receptor shall not exceed 3.34x10-3 percent of the entrance AKR, at a distance of 10 cm from any accessible surface of the fluoroscopic imaging assembly beyond the plane of the image receptor. Radiation therapy simulation systems shall be exempt from this requirement provided the systems are intended only for remote control operation.
5.1.2 Measuring compliance. The AKR shall be measured in accordance with 5.3.2. The AKR due to transmission through the primary barrier combined with radiation from the fluoroscopic image receptor shall be determined by measurements averaged over an area of 100 square cm with no linear dimension greater than 20 cm. If the source is below the tabletop, the measurement shall be made with the input surface of the fluoroscopic imaging assembly positioned 30 cm above the tabletop. If the source is above the tabletop and the SID is variable, the measurement shall be made with the end of the beam-limiting device or spacer as close to the tabletop as it can be placed, provided that it shall not be closer than 30 cm. Movable grids and compression devices shall be removed from the useful beam during the measurement. For all measurements, the attenuation block shall be positioned in the useful beam 10 cm from the point of measurement of entrance AKR and between this point and the input surface of the fluoroscopic imaging assembly.
5.2 Field Limitation.
5.2.1 Angulation. For fluoroscopic equipment manufactured after February 25, 1978, when the angle between the image receptor and the beam axis of the x-ray beam is variable, means shall be provided to indicate when the axis of the x-ray beam is perpendicular to the plane of the image receptor. Compliance with 5.2.4 and 5.2.5 shall be determined with the beam axis indicated to be perpendicular to the plane of the image receptor.
5.2.2 Further means for limitation. Means shall be provided to permit further limitation of the x-ray field to sizes smaller than the limits of 5.2.4 and 5.2.5. Beam-limiting devices manufactured after May 22, 1979, and incorporated in equipment with a variable SID and/or capability of a visible area of greater than 300 square cm, shall be provided with means for stepless adjustment of the x-ray field. Equipment with a fixed SID and the capability of a visible area of no greater than 300 square cm shall be provided with either stepless adjustment of the x-ray field or with a means to further limit the x-ray field size at the plane of the image receptor to 125 square cm or less. Stepless adjustment shall, at the greatest SID, provide continuous field sizes from the maximum obtainable to a field size containable in a square of 5 cm by 5 cm. This paragraph does not apply to non-image-intensified fluoroscopy.
5.2.3 Non-image-intensified fluoroscopy. The x-ray field produced by non-image-intensified fluoroscopic equipment shall not extend beyond the entire visible area of the image receptor. Means shall be provided for stepless adjustment of field size. The minimum field size, at the greatest SID, shall be containable in a square of 5 cm by 5 cm.
5.2.4 Fluoroscopy and radiography using the fluoroscopic imaging assembly with inherently circular image receptors.
5.2.4.1 For fluoroscopic equipment manufactured before June 10, 2006, other than radiation therapy simulation systems, the following applies:
5.2.4.1.1 Neither the length nor width of the x-ray field in the plane of the image receptor shall exceed that of the visible area of the image receptor by more than 3 percent of the SID. The sum of the excess length and the excess width shall be no greater than 4 percent of the SID.
5.2.4.1.2 For rectangular x-ray fields used with circular image receptors, the error in alignment shall be determined along the length and width dimensions of the x-ray field which pass through the center of the visible area of the image receptor.
5.2.4.2 For fluoroscopic equipment manufactured on or after June 10, 2006, other than radiation simulation systems, the maximum area of the x-ray field in the plane of the image receptor shall conform with one of the following requirements:
5.2.4.2.1 When any linear dimension of the visible area of the image receptor measured through the center of the visible area is less than or equal to 34 cm in any direction, at least 80 percent of the area of the x-ray field overlaps the visible area of the image receptor, or
5.2.4.2.2 When any linear dimension of the visible area of the image receptor measured through the center of the visible area is greater than 34 cm in any direction, the x-ray field measured along the direction of greatest misalignment with the visible area of the image receptor does not extend beyond the edge of the visible area of the image receptor by more than 2 cm.
5.2.5 Fluoroscopy and radiography using fluoroscopic imaging assembly with inherently rectangular image receptors. For x-ray systems manufactured on or after June 10, 2006, the following applies:
5.2.5.1 Neither the length nor width of the x-ray field in the plane of the image receptor shall exceed that of the visible area of the image receptor by more than 3 percent of the SID. The sum of the excess length and the excess width shall be no greater than 4 percent of the SID.
5.2.5.2 The error in alignment shall be determined along the length and width dimensions of the x-ray field which pass through the center of the visible area of the image receptor.
5.3 If the fluoroscopic x-ray field size is adjusted automatically as the SID or image receptor size is changed, a capability may be provided for overriding the automatic adjustment in case of system failure. If it is so provided, a signal visible at the fluoroscopist’s position shall indicate whenever the automatic field adjustment is overridden. Each such system failure override switch shall be clearly labeled as follows:
FOR X-RAY FIELD LIMITATION SYSTEM FAILURE
5.3.1 Activation of Tube. X-ray production in the fluoroscopic mode shall be controlled by a device which requires continuous pressure by the operator for the entire time of any exposure. When recording serial radiographic images from the fluoroscopic image receptor, the operator shall be able to terminate the x-ray exposure(s) at any time, but means may be provided to permit completion of any single exposure of the series in process.
5.3.2 Air Kerma Rates. For fluoroscopic equipment, the following requirements apply:
5.4 Fluoroscopic equipment manufactured before May 19, 1995.
5.4.1 Equipment provided with automatic exposure rate control (AERC) shall not be operable at any combination of tube potential and current that will result in an AKR in excess of 88 mGy per minute (vice 10 R/min exposure rate) at the measurement point specified in 5.6, except as specified in 5.4.4.1.
5.4.2 Equipment provided without AERC shall not be operable at any combination of tube potential and current that will result in an AKR in excess of 44 mGy per minute (vice 5 R/min exposure rate) at the measurement point specified in 5.6, except as specified in 5.4.4.1.
5.4.3 Equipment provided with both an AERC mode and a manual mode shall not be operable at any combination of tube potential and current that will result in an AKR in excess of 88 mGy per minute (vice 10 R/min exposure rate) in either mode at the measurement point specified in 5.6, except as specified in 5.4.4.1.
5.4.4 Equipment may be modified in accordance with this Part to comply with 5.5. When the equipment is modified, it shall bear a label indicating the date of the modification and the statement:
5.4.4.1 Exceptions:
5.4.4.1.1 During recording of fluoroscopic images, or
5.4.4.1.2 When a mode of operation has an optional high-level control, in which case that mode shall not be operable at any combination of tube potential and current that will result in an AKR in excess of the rates specified in paragraphs (1), (2) and (3) at the measurement point specified in 5.6, unless the high-level control is activated. Special means of activation of high-level controls shall be required. The high-level control shall be operable only when continuous manual activation is provided by the operator. A continuous signal audible to the fluoroscopist shall indicate that the high-level control is being employed.
5.5 Fluoroscopic equipment manufactured on or after May 19, 1995.
5.5.1 Shall be equipped with AERC if operable at any combination of tube potential and current that results in an AKR greater than 44 mGy per minute (vice 5 R/min exposure rate) at the measurement point specified in 5.6. Provision for manual selection of technique factors may be provided.
5.5.2 Shall not be operable at any combination of tube potential and current that will result in an AKR in excess of 88 mGy per minute (vice 10 R/min exposure rate) at the measurement point specified in 5.6, except as specified in 5.5.3.
5.5.3 Exceptions:
5.5.3.1 For equipment manufactured prior to June 10, 2006, during the recording of images from a fluoroscopic image receptor using photographic film or a video camera when the x-ray source is operated in a pulsed mode.
5.5.3.2 For equipment manufactured on or after June 10, 2006, during the recording of images from the fluoroscopic image receptor for the purpose of providing the user with a recorded image(s) after termination of the exposure. Such recording does not include images resulting from a last-image-hold feature that are not recorded.
5.5.3.3 When a mode of operation has an optional high-level control and the control is activated, in which case the equipment shall not be operable at any combination of tube potential and current that will result in an AKR in excess of 176 mGy per minute (vice 20 R/min exposure rate) at the measurement point specified in 5.6. Special means of activation of high-level controls shall be required. The high-level control shall be operable only when continuous manual activation is provided by the operator. A continuous signal audible to the fluoroscopist shall indicate that the high-level control is employed.
5.6 Measuring compliance. Compliance with this subsection shall be determined as follows:
5.6.1 If the source is below the x-ray table, the AKR shall be measured at 1 cm above the tabletop or cradle.
5.6.2 If the source is above the x-ray table, the AKR shall be measured at 30 cm above the tabletop with the end of the beam-limiting device or spacer positioned as closely as possible to the point of measurement.
5.6.3 In a C-arm type of fluoroscope, the AKR shall be measured at 30 cm from the input surface of the fluoroscopic imaging assembly, with the source positioned at any available SID, provided that the end of the beam-limiting device or spacer is no closer than 30 cm from the input surface of the fluoroscopic imaging assembly.
5.6.4 In a C-arm type of fluoroscope having an SID less than 45 cm, the AKR shall be measured at the minimum SSD.
5.6.5 In a lateral type of fluoroscope, the air kerma rate shall be measured at a point 15 cm from the centerline of the x-ray table and in the direction of the x-ray source with the end of the beam-limiting device or spacer positioned as closely as possible to the point of measurement. If the tabletop is movable, it shall be positioned as closely as possible to the lateral x-ray source, with the end of the beam-limiting device or spacer no closer than 15 cm to the centerline of the x-ray table.
5.7 Exemptions. Fluoroscopic radiation therapy simulation systems are exempt from the requirements set forth in this subsection when used for therapy simulation purposes.
5.8 Reserved.
5.9 Indication of potential and current. During fluoroscopy and cinefluorography, x-ray tube potential and current shall be continuously indicated. Deviation of x-ray tube potential and current from the indicated value shall not exceed the maximum deviation as stated by the manufacturer.
5.10 Source-skin distance.
5.10.1 Means shall be provided to limit the source-skin distance to not less than 38 cm on stationary fluoroscopes and to not less than 30 cm on mobile and portable fluoroscopes. In addition, for fluoroscopes intended for specific surgical application that would be prohibited at the source-skin distances specified in this paragraph, provisions may be made for operating at shorter source-skin distances but in no case less than 20 cm.
5.10.2 For stationary, mobile, or portable C-arm fluoroscopic systems manufactured on or after June 10, 2006, having a maximum source-image receptor distance of less than 45 cm, means shall be provided to limit the source-skin distance to not less than 19 cm. Such systems shall be labeled for extremity use only. In addition, for those systems intended for specific surgical application that would be prohibited at the source-skin distance specified in this paragraph, provisions may be made for operation at shorter source-skin distances but in no case less than 10 cm.
5.11 Fluoroscopic irradiation time, display, and signal.
5.11.1 Fluoroscopic equipment manufactured before June 10, 2006:
5.11.1.1 Shall be provided with means to preset the cumulative irradiation time of the fluoroscopic tube. The maximum cumulative time of the timing device shall not exceed 5 minutes without resetting. A signal audible to the fluoroscopist shall indicate the completion of any preset cumulative irradiation time. Such signal shall continue to sound while x-rays are produced until the timing device is reset. Fluoroscopic equipment may be modified in accordance with 21 CFR 1020.30(q) to comply with the requirements of this paragraph. When the equipment is modified, it shall bear a label indicating the statement:
5.12 Modified to comply with 21 CFR 1020.32(h)(2)
5.12.1 As an alternative to the requirements of this paragraph, radiation therapy simulation systems may be provided with a means to indicate the total cumulative exposure time during which x-rays were produced, and which is capable of being reset between x-ray examinations.
5.13 For x-ray controls manufactured on or after June 10, 2006, there shall be provided for each fluoroscopic tube:
5.13.1 A display of the fluoroscopic irradiation time at the fluoroscopist’s working position. This display shall function independently of the audible signal described in this subsection. The following requirements apply:
5.13.1.1 When the x-ray tube is activated, the fluoroscopic irradiation time in minutes and tenths of minutes shall be continuously displayed and updated at least once every 6 seconds.
5.13.1.2 The fluoroscopic irradiation time shall also be displayed within 6 seconds of termination of an exposure and remain displayed until reset.
5.13.1.3 Means shall be provided to reset the display to zero prior to the beginning of a new examination or procedure.
5.13.2 A signal audible to the fluoroscopist shall sound for each passage of 5 minutes of fluoroscopic irradiation time during an examination or procedure. The signal shall sound until manually reset or, if automatically reset, for at least 2 seconds.
5.14 Mobile and portable fluoroscopes. In addition to the other requirements of this subsection, mobile and portable fluoroscopes shall provide an image receptor incorporating more than a simple fluorescent screen.
5.15 Display of last-image-hold (LIH). Fluoroscopic equipment manufactured on or after June 10, 2006, shall be equipped with means to display LIH image following termination of the fluoroscopic exposure.
5.16 For an LIH image obtained by retaining pretermination fluoroscopic images, if the number of images and method of combining images are selectable by the user, the selection shall be indicated prior to initiation of the fluoroscopic exposure.
5.17 For an LIH image obtained by initiating a separate radiographic-like exposure at the termination of fluoroscopic imaging, the technique factors for the LIH image shall be selectable prior to the fluoroscopic exposure, and the combination selected shall be indicated prior to initiation of the fluoroscopic exposure.
5.18 Means shall be provided to clearly indicate to the user whether a displayed image is the LIH radiograph or fluoroscopy. Display of the LIH radiograph shall be replaced by the fluoroscopic image concurrently with re-initiation of fluoroscopic exposure, unless separate displays are provided for the LIH radiograph and fluoroscopic images.
5.19 Displays of values of AKR and cumulative air kerma. Fluoroscopic equipment manufactured on or after June 10, 2006, shall display at the fluoroscopist’s working position the AKR and cumulative air kerma. The following requirements apply for each x-ray tube used during an examination or procedure:
5.20 When the x-ray tube is activated and the number of images produced per unit time is greater than six images per second, the AKR in mGy/min shall be continuously displayed and updated at least once every second.
5.21 The cumulative air kerma in units of mGy shall be displayed either within 5 seconds of termination of an exposure or displayed continuously and updated at least once every 5 seconds.
5.22 The display of the AKR shall be clearly distinguishable from the display of the cumulative air kerma.
5.23 The AKR and cumulative air kerma shall represent the value for conditions of free-in-air irradiation at one of the following reference locations specified according to the type of fluoroscope.
5.23.1 For fluoroscopes with x-ray source below the x-ray table, x-ray source above the table, or of lateral type, the reference location shall be the respective locations specified in 5.6.1, 5.6.2 or 5.6.5.
5.23.2 For C-arm fluoroscopes, the reference location shall be 15 cm from the isocenter toward the x-ray source along the beam axis. Alternatively, the reference location shall be at a point specified by the manufacturer to represent the location of the intersection of the x-ray beam with the patient’s skin.
5.24 Means shall be provided to reset to zero the display of cumulative air kerma prior to the commencement of a new examination or procedure.
5.25 The displayed AKR and cumulative air kerma shall not deviate from the actual values by more than ±35 percent over the range of 6 mGy/min and 100 mGy to the maximum indication of AKR and cumulative air kerma, respectively. Compliance shall be determined with an irradiation time greater than 3 seconds.
5.26 Control of Scattered Radiation.
5.26.1 Fluoroscopic table designs when combined with procedures utilized shall be such that no unprotected part of any staff or ancillary individual's body shall be exposed to unattenuated scattered radiation which originates from under the table. The attenuation required shall be not less than 0.25 millimeter lead equivalent.
5.26.2 Equipment configuration when combined with procedures shall be such that no portion of any staff or ancillary individual's body, except the extremities, shall be exposed to the unattenuated scattered radiation emanating from above the tabletop unless that individual:
5.26.2.1 Is at least 120 centimeters from the center of the useful beam; or
5.26.2.2 The radiation has passed through not less than 0.25 millimeter lead equivalent material including, but not limited to, drapes, Bucky-slot cover panel, or self-supporting curtains, in addition to any lead equivalency provided by the protective apron referred to in 3.1.5.2.
5.27 The Agency may grant exemptions to 5.26.2. where a sterile field will not permit the use of the normal protective barriers. Where the use of prefitted sterilized covers for the barriers is practical, the Agency shall not permit such exemption. See Appendix C for a suggested list of fluoroscopic procedures where such exemptions will be automatically granted.
5.28 Operator Qualifications.
5.28.1 The facility shall ensure that only a licensed practitioner of the healing arts or a radiologic technologist be allowed to operate fluoroscopic x-ray systems. A licensed practitioner includes any health practitioner of the healing Arts who is licensed in the state to diagnose and treat individuals and who operates within the scope defined in the state law. This includes physicians and physician assistants and excludes nurse practitioners.
5.28.2 All persons operating fluoroscopic x-ray systems shall have completed at least the following training before using fluoroscopy independently: 5.28.1.
5.28.2.1 Biological effects of x-ray;
5.28.2.2 Principles of radiation protection;
5.28.2.3 Factors affecting fluoroscopic outputs;
5.28.2.4 Dose reduction techniques for fluoroscopic x-ray systems;
5.28.2.5 Principles and operation of the specific fluoroscopic x-ray system(s) to be used;
5.28.2.6 Fluoroscopic and fluorographic outputs of each mode of operation on the system(s) to be used clinically; and
5.28.2.7 Applicable requirements of these regulations.
5.29 The facility shall either provide annual in-service training for all operators of fluoroscopic x-ray systems used for high dose, high risk procedures, or require evidence of annual continuing medical education in fluoroscopic radiation safety and patient dose management.
5.30 Documentation pertaining to the requirements of 5.28.2 and 5.29, shall be maintained for review for three years.
5.31 Equipment Operation.
5.31.1 All imaging formed by the use of fluoroscopic x-ray systems shall be viewed, directly or indirectly, and interpreted by a licensed practitioner of the healing arts.
5.31.2 The operation of fluoroscopic x-ray systems by radiologic technologists [or equivalent] shall be performed under the direct supervision of a licensed practitioner of the healing arts who meets the requirements of 5.28.
5.31.3 Radiologic technology students shall not be allowed to operate fluoroscopic x-ray systems unless directly supervised by a licensed practitioner of the healing arts or radiologic technologist as specified in 5.28.1.
5.31.4 Overhead fluoroscopy shall not be used as a positioning tool for general purpose radiographic examinations.
5.31.5 Facilities shall maintain a record of the cumulative fluoroscopic exposure time used and the number of fluorographic images recorded for each examination. This record shall include patient identification, type and date of examination, the fluoroscopic system used, and operator's name.
6.1 Control and indication of technique factors.
6.1.1 Visual indication. The technique factors to be used during an exposure shall be indicated before the exposure begins, except when automatic exposure controls are used, in which case the technique factors which are set prior to the exposure shall be indicated. On equipment having fixed technique factors, this requirement may be met by permanent markings. Indication of technique factors shall be visible from the operator’s position except in the case of spot films made by the fluoroscopist.
6.1.2 Timers. Means shall be provided to terminate the exposure at a preset time interval, a preset product of current and time, a preset number of pulses, or a preset radiation exposure to the image receptor.
6.1.2.1 Except during serial radiography, the operator shall be able to terminate the exposure at any time during an exposure of greater than one-half second. Except during panoramic dental radiography, termination of exposure shall cause automatic resetting of the timer to its initial setting or to zero. It shall not be possible to make an exposure when the timer is set to a zero or off position if either position is provided.
6.1.2.2 During serial radiography, the operator shall be able to terminate the x-ray exposure(s) at any time, but means may be provided to permit completion of any single exposure of the series in process.
6.2 Automatic exposure controls. When an automatic exposure control is provided:
6.2.1 Indication shall be made on the control panel when this mode of operation is selected;
6.2.2 When the x-ray tube potential is equal to or greater than 51 kilovolts peak (kVp), the minimum exposure time for field emission equipment rated for pulse operation shall be equal to or less than a time interval equivalent to two pulses and the minimum exposure time for all other equipment shall be equal to or less than 1/60 second or a time interval required to deliver 5 milliampere-seconds (mAs), whichever is greater;
6.2.3 Either the product of peak x-ray tube potential, current, and exposure time shall be limited to not more than 60 kilowatt-seconds (kWs) per exposure or the product of x-ray tube current and exposure time shall be limited to not more than 600 mAs per exposure, except when the x-ray tube potential is less than 51 kVp, in which case the product of x-ray tube current and exposure time shall be limited to not more than 2,000 mAs per exposure; and
6.2.4 A visible signal shall indicate when an exposure has been terminated at the limits described in 6.2.3, and manual resetting shall be required before further automatically timed exposures can be made.
6.2.5 Accuracy. Deviation of technique factors from indicated values shall not exceed the limits given by the manufacturer.
6.2.5.1 Reproducibility. The following requirements shall apply when the equipment is operated on an adequate power supply as specified by the manufacturer:
6.2 Coefficient of variation. For any specific combination of selected technique factors, the estimated coefficient of variation of the air kerma shall be no greater than 0.05.
6.3 Measuring compliance. Determination of compliance shall be based on 10 consecutive measurements taken within a time period of 1 hour. Equipment manufactured after September 5, 1978, shall be subject to the additional requirement that all variable controls for technique factors shall be adjusted to alternate settings and reset to the test setting after each measurement. The percent line-voltage regulation shall be within ±1 of the mean value for all measurements. For equipment having automatic exposure controls, compliance shall be determined with a sufficient thickness of attenuating material in the useful beam such that the technique factors can be adjusted to provide individual exposures of a minimum of 12 pulses on field emission equipment rated for pulsed operation or no less than one-tenth second per exposure on all other equipment.
6.4 Linearity. The following requirements apply when the equipment is operated on a power supply as specified by the manufacturer in accordance with 21 CFR Part 1020 for any fixed x-ray tube potential within the range of 40 percent to 100 percent of the maximum rated.
6.4.1 Equipment having independent selection of x-ray tube current (mA). The average ratios of air kerma to the indicated milliampere-seconds product (mGy/mAs) obtained at any two consecutive tube current settings shall not differ by more than 0.10 times their sum. This is: ׀X1 – X2׀ ≤ 0.10(X1 + X2); where X1 and X2 are the average mGy/mAs values obtained at each of two consecutive mAs selector settings or at two settings differing by no more than a factor of 2 where the mAs selector provides continuous selection.
6.4.2 Equipment having selection of x-ray tube current-exposure time product (mAs). For equipment manufactured after May 3, 1994, the average ratios of air kerma to the indicated milliampere-seconds product (mGy/mAs) obtained at any two consecutive mAs selector settings shall not differ by more than 0.10 times their sum. This is: ׀X1 – X2׀ ≤ 0.10(X1 + X2); where X1 and X2 are the average mGy/mAs values obtained at each of two consecutive mAs selector settings or at two settings differing by no more than a factor of 2 where the mAs selector provides continuous selection.
6.4.3 Measuring compliance. Determination of compliance will be based on 10 exposures, made within 1 hour, at each of the two settings. These two settings may include any two focal spot sizes except where one is equal to or less than 0.45 mm and the other is greater than 0.45 mm. For purposes of this requirement, focal spot size is the focal spot size specified by the x-ray tube manufacturer. The percent line-voltage regulation shall be determined for each measurement. All values for percent line-voltage regulation at any one combination of technique factors shall be within ±1 of the mean value for all measurements at these technique factors.
6.5 Field limitation and alignment for mobile, portable, and stationary general purpose x-ray systems. Except when spot-film devices are in service, mobile, portable, and stationary general purpose radiographic x-ray systems shall meet the following requirements:
6.6 Variable x-ray field limitation. A means for stepless adjustment of the size of the x-ray field shall be provided. Each dimension of the minimum field size at an SID of 100 cm shall be equal to or less than 5 cm.
6.6.1 Visual definition.
6.6.1.1 Means for visually defining the perimeter of the x-ray field shall be provided. The total misalignment of the edges of the visually defined field with the respective edges of the x-ray field along either the length or width of the visually defined field shall not exceed 2 percent of the distance from the source to the center of the visually defined field when the surface upon which it appears is perpendicular to the axis of the x-ray beam.
6.6.1.2 When a light localizer is used to define the x-ray field, it shall provide an average illuminance of not less than 160 lux (15 footcandles) at 100 cm or at the maximum SID, whichever is less. The average illuminance shall be based on measurements made in the approximate center of each quadrant of the light field. Radiation therapy simulation systems are exempt from this requirement.
6.6.1.3 The edge of the light field at 100 cm or at the maximum SID, whichever is less, shall have a contrast ratio, corrected for ambient lighting, of not less than 4 in the case of beam-limiting devices designed for use on stationary equipment, and a contrast ratio of not less than 3 in the case of beam-limiting devices designed for use on mobile and portable equipment. The contrast ratio is defined as I1/I2, where I1 is the illuminance 3 mm from the edge of the light field toward the center of the field; and I2 is the illuminance 3 mm from the edge of the light field away from the center of the field. Compliance shall be determined with a measuring aperture of 1 mm.
6.7 Field indication and alignment on stationary general purpose x-ray equipment. Except when spot-film devices are in service, stationary general purpose x-ray systems shall meet the following requirements in addition to those prescribed in 6.5.
6.7.1 Means shall be provided to indicate when the axis of the x-ray beam is perpendicular to the plane of the image receptor, to align the center of the x-ray field with respect to the center of the image receptor to within 2 percent of the SID, and to indicate the SID to within 2 percent;
6.7.2 The beam-limiting device shall numerically indicate the field size in the plane of the image receptor to which it is adjusted;
6.7.3 Indication of field size dimensions and SIDs shall be specified in centimeters and/or inches and shall be such that aperture adjustments result in x-ray field dimensions in the plane of the image receptor which correspond to those indicated by the beam-limiting device to within 2 percent of the SID when the beam axis is indicated to be perpendicular to the plane of the image receptor; and
6.7.4 Compliance measurements will be made at discrete SIDs and image receptor dimensions in common clinical use (such as SIDs of 100, 150, and 200 cm and/or 36, 40, 48, 72 inches and nominal image receptor dimensions of 13, 18, 24, 30, 35, 40, and 43 cm and/or 5, 7, 8, 9, 10, 11, 12, 14, and 17 inches) or at any other specific dimensions at which the beam-limiting device or its associated diagnostic x-ray system is uniquely designed to operate.
6.8 Field limitation on radiographic x-ray equipment other than general purpose radiographic systems.
6.8.1 Equipment for use with intraoral image receptors. Radiographic equipment designed for use with an intraoral image receptor shall be provided with means to limit the x-ray beam such that:
6.8.1.1 If the minimum source-to-skin distance (SSD) is 18 cm or more, the x-ray field at the minimum SSD shall be containable in a circle having a diameter of no more than 7 cm; and
6.8.1.2 If the minimum SSD is less than 18 cm, the x-ray field at the minimum SSD shall be containable in a circle having a diameter of no more than 6 cm.
6.9 X-ray systems designed for one image receptor size. Radiographic equipment designed for only one image receptor size at a fixed SID shall be provided with means to limit the field at the plane of the image receptor to dimensions no greater than those of the image receptor, and to align the center of the x-ray field with the center of image receptor to within 2 percent of the SID, or shall be provided with means to both size and align the x-ray field such that the x-ray field at the plane of the image receptor does not extend beyond the edge of the image receptor.
6.10 Systems designed for mammography.
6.10.1 Radiographic systems designed only for mammography and general purpose radiography systems, when special attachments for mammography are in service, manufactured on or after November 1, 1977, and before September 30, 1999, shall be provided with means to limit the useful beam such that the x-ray field at the plane of the image receptor does not extend beyond any edge of the image receptor at any designated SID except the edge of the image receptor designed to be adjacent to the chest wall where the x-ray field may not extend beyond this edge by more than 2 percent of the SID. This requirement can be met with a system that performs as prescribed in 6.11.When the beam-limiting device and image receptor support device are designed to be used to immobilize the breast during a mammographic procedure and the SID may vary, the SID indication specified in 6.11 shall be the maximum SID for which the beam-limiting device or aperture is designed.
6.10.2 Mammographic beam-limiting devices manufactured on or after September 30, 1999, shall be provided with a means to limit the useful beam such that the x-ray field at the plane of the image receptor does not extend beyond any edge of the image receptor by more than 2 percent of the SID. This requirement can be met with a system that performs as prescribed in 6.11.For systems that allow changes in SID, the SID indication specified in 6.11 shall be the maximum SID for which the beam-limiting deviced or aperture is designed.
6.10.3 Each image receptor support device manufactured on or after November 1, 1977, intended for installation on a system designed for mammography shall have clear and permanent markings to indicate the maximum image receptor size for which it is designed.
6.11 Other x-ray systems. Radiographic systems not specifically covered in 6.5, 6.7, 6.8.1.2, 6.10, 6.18, and systems covered in 6.8.1, which are also designed for use with extraoral image receptors and when used with an extraoral image receptor, shall be provided with means to limit the x-ray field in the plane of the image receptor so that such field does not exceed each dimension of the image receptor by more than 2 percent of the SID, when the axis of the x-ray beam is perpendicular to the plane of the image receptor. In addition, means shall be provided to align the center of the x-ray field with the center of the image receptor to within 2 percent of the SID, or means shall be provided to both size and alignment the x-ray field such that the x-ray field at the plane of the image receptor does not extend beyond any edge of the image receptor. These requirements may be met with:
6.11.1 A system which performs in accordance with 6.5 and 6.7; or when alignment means are also provided, may be met with either;
6.11.2 An assortment of removable, fixed-aperture, beam-limiting devices sufficient to meet the requirement for each combination of image receptor size and SID for which the unit is designed. Each such device shall have clear and permanent markings to indicate the image receptor size and SID for which it is designed; or
6.11.3 A beam-limiting device having multiple fixed apertures sufficient to meet the requirement for each combination of image receptor size and SID for which the unit is designed. Permanent, clearly legible markings shall indicate the image receptor size and SID for which each aperture is designed and shall indicate which aperture is in position for use.
6.12 Positive beam limitation (PBL). The requirements of this subsection shall apply to radiographic systems which contain PBL.
6.12.1 Field size. When a PBL system is provided, it shall prevent x-ray production when:
6.12.1.1 Either the length or width of the x-ray field in the plane of the image receptor differs from the corresponding image receptor dimension by more than 3 percent of the SID; or
6.12.1.2 The sum of the length and width differences stated in 6.12.1.1 without regard to sign exceeds 4 percent of the SID.
6.13.1.3 The beam-limiting device is at an SID for which PBL is not designed for sizing.
6.13 Conditions for PBL. When provided, the PBL system shall function as described in 6.12.1 whenever all the following conditions are met:
6.13.1 The image receptor is inserted into a permanently mounted cassette holder;
6.13.2 The image receptor length and width are less than 50 cm;
6.13.3 The x-ray beam axis is within ±3 degrees of vertical and the SID is 90 cm to 130 cm inclusive; or the x-ray beam axis is within ±3 degrees of horizontal and the SID is 90 cm to 205 cm inclusive;
6.13.4 The x-ray beam axis is perpendicular to the plane of the image receptor to within ±3 degrees; and
6.13.5 Neither tomographic nor stereoscopic radiography is being performed.
6.14 Measuring compliance. Compliance with the requirements of 6.12.1. shall be determined when the equipment indicates that the beam axis is perpendicular to the plane of the image receptor and the provisions of 6.13 are met. Compliance shall be determined no sooner than 5 second after insertion of the image receptor.
6.15 Operator initiated undersizing. The PBL system shall be capable of operating such that, at the discretion of the operator, the size of the field may be made smaller than the size of the image receptor through stepless adjustment of the field size. Each dimension of the minimum field size at an SID of 100 cm shall be equal to or less than 5 cm. Return to PBL function as described in 6.12.1 shall occur automatically upon any change of image receptor size or SID.
6.16 Override of PBL. A capability may be provided for overriding PBL in case of system failure and for servicing the system. This override may be for all SIDs and image receptor sizes. A key shall be required for any override capability that is accessible to the operator. It shall not be possible to remove the key while PBL is overridden. Each such key switch or key shall be clearly and durably labeled as follows:
6.17 For X-Ray Field Limitation System Failure
6.17.1 The override capability is considered accessible to the operator if it is referenced in the operator’s manual or in other material intended for the operator or if its location is such that the operator would consider it part of the operational controls.
6.18 Field limitation and alignment for spot-film devices. The following requirements shall apply to spot-film devices, except when the spot-film device is provided for use with a radiation therapy simulation system:
6.18.1 Means shall be provided between the source and the patient for adjustment of the x-ray field size in the plane of the image receptor to the size of that portion of the image receptor which has been selected on the spot-film selector. Such adjustment shall be accomplished automatically when the x-ray field size in the plane of the image receptor is greater than the selected portion of the image receptor. If the x-ray field size is less than the size of the selected portion of the image receptor, the field size shall not open automatically to the size of the selected portion of the image receptor unless the operator has selected that mode of operation.
6.18.2 Neither the length nor width of the x-ray field in the plane of the image receptor shall differ from the corresponding dimensions of the selected portion of the image receptor by more than 3 percent of the SID when adjusted for full coverage of the selected portion of the image receptor. The sum, without regard to sign, of the length and width differences shall not exceed 4 percent of the SID. On spot film devices manufactured after February 25, 1978, if the angle between the plane of the image receptor and beam axis is variable, means shall be provided to indicate when the axis of the x-ray beam is perpendicular to the plane of the image receptor, and compliance shall be determined with the beam axis indicated to be perpendicular to the plane of the image receptor.
6.18.3 The center of the x-ray field in the plane of the image receptor shall be aligned with the center of the selected portion of the image receptor to within 2 percent of the SID.
6.18.4 Means shall be provided to reduce the x-ray field size in the plane of the image receptor to a size smaller than the selected portion of the image receptor such that:
6.18.4.1 For spot-film devices used on fixed-SID fluoroscopic systems which are not required to, and do not provide stepless adjustment of the x-ray field, the minimum field size, at the greatest SID, does not exceed 125 square cm; or
6.18.4.2 For spot-film devices used on fluoroscopic systems that have a variable SID and/or stepless adjustment of the field size, the minimum field size, at the greatest SID, shall be containable in a square of 5 cm by 5 cm.
6.19 A capability may be provided for overriding the automatic x-ray field size adjustment in case of system failure. If it is so provided, a signal visible at the fluoroscopist’s position shall indicate whenever the automatic x-ray field size adjustment override is engaged. Each such system failure override switch shall be clearly labeled as follows:
6.20 For X-ray Field Limitation System Failure
6.20.1 Source-skin distance.
6.20.1.1 X-ray systems designed for use with an intraoral image receptor shall be provided with means to limit the source-skin distance to not less than:
6.20.1.1.1 Eighteen cm if operable above 50 kVp; or
6.20.1.1.2 Ten cm if not operable above 50 kVp.
6.21 Mobile and portable x-ray systems other than dental shall be provided with means to limit the source-skin distance to not less than 30 cm. (See Appendix B for dental systems)
6.22 Beam-on indicators. The x-ray control shall provide visual indication whenever x-rays are produced. In addition, a signal audible to the operator shall indicate that the exposure has terminated.
6.23 Reserved.
6.24 Radiation from capacitor energy storage equipment. Radiation emitted from the x-ray tube shall not exceed:
6.24.1 An air kerma of 0.26 microGy (vice 0.03 mR exposure) in 1 minute at 5 cm from any accessible surface of the diagnostic source assembly, with the beam-limiting device fully open, the system fully charged, and the exposure switch, timer, or any discharge mechanism not activated. Compliance shall be determined by measurements averaged over an area of 100 square cm, with no linear dimensions greater than 20 cm: and
6.24.2 An air kerma of 0.88 mGy (vice 100 mR exposure) in one hour at 100 cm from the x-ray source, with beam-limiting device fully open, when the system is discharged through the x-ray tube either manually or automatically by use of a discharge switch or deactivation of the input power. Compliance shall be determined by measurements of the maximum air kerma per discharge multiplied by the total number of discharges in 1 hour (duty cycle). The measurements shall be averaged over an area of 100 square cm with no linear dimension greater than 20 cm.
6.25 Primary protective barrier for mammography x-ray systems.
6.25.1 For x-ray systems manufactured after September 5, 1978, and before September 30, 1999, which are designed only for mammography, the transmission of the primary beam through any image receptor support provided with the system shall be limited such that the air kerma 5 cm from any accessible surface beyond the plane of the image receptor supporting device does not exceed 0.88 microGy (vice 0.1 mR exposure) for each activation of the tube.
6.25.2 For mammographic x-ray systems manufactured on or after September 30, 1999:
6.25.2.1 At any SID where exposures can be made, the image receptor support device shall provide a primary protective barrier that intercepts the cross section of the useful beam along every direction except at the chest wall edge.
6.25.2.2 The x-ray system shall not permit exposure unless the appropriate barrier is in place to intercept the useful beam as required in 6.25.2.1.
6.25.2.3 The transmission of the useful beam through the primary protective barrier shall be limited such that the air kerma 5 cm from any accessible surface beyond the plane of the primary protective barrier does not exceed 0.88 microGy (vice 0.1 mR exposure) for each activation of the tube.
6.26 Compliance with the requirements of 6.25.1 and 6.25.2.3 for transmission shall be determined with the x-ray system operated at the minimum SID for which it is designed, at maximum rated peak tube potential, at the maximum rated product of x-ray tube current and exposure time (mAs) for the maximum rated peak tube potential, and by measurements averaged over an area of 100 square cm with no linear dimension greater than 20 cm. The sensitive volume of the radiation measuring instrument shall not be positioned beyond the edge of the primary protective barrier along the chest wall side.
6.27 Beam Limitation, Except Mammographic Systems. The useful beam shall be limited to the area of clinical interest. This shall be deemed to have been met if a positive beam limiting device meeting manufacturer's specifications and the requirements of 6.12 have been properly used or if evidence of collimation is shown on at least three sides or three corners of the film (for example, projections from the shutters of the collimator, cone cutting at the corners, or borders at the film's edge).
6.28 Radiation Exposure Control.
6.28.1 Exposure Initiation. Means shall be provided to initiate the radiation exposure by a deliberate action on the part of the operator, such as the depression of a switch. Radiation exposure shall not be initiated without such an action. In addition, it shall not be possible to initiate an exposure when the timer is set to a "zero" or "off" position if either position is provided.
6.28.2 Exposure Indication. Means shall be provided for visual indication observable at or from the operator's protected position whenever x-rays are produced. In addition, a signal audible to the operator shall indicate that the exposure has terminated.
6.28.3 Operator Protection, Except Veterinary Systems.
6.28.3.1 Stationary Systems. Stationary x-ray systems shall be required to have the x-ray control permanently mounted in a protected area so that the operator is required to remain in that protected area during the entire exposure.
6.28.3.2 Mobile and Portable Systems. Mobile and portable x-ray systems which are:
6.28.3.2.1 Used continuously for greater than one week in the same location, i.e., a room or suite, shall meet the requirements of 6.28.3.1
6.28.3.2.2 Used for less than one week at the same location shall be provided with either a protective barrier at least 2 meters (6.5 feet) high for operator protection during exposures, or means shall be provided to allow the operator to be at least 2.7 meters (9 feet) from the tube housing assembly during the exposure.
6.29 Operator Protection for Veterinary Systems. All stationary, mobile or portable x-ray systems used for veterinary work shall be provided with either a 2 meter (6.5 feet) high protective barrier for operator protection during exposures, or shall be provided with means to allow the operator to be at least 2.7 meters (9 feet) from the tube housing assembly during exposures. Refer to Appendix B for hand-held intraoral dental radiographic units used in veterinary practice.
6.30 Tube Stands for Portable X-Ray Systems. A tube stand or other mechanical support shall be used for portable x-ray systems, so that the x-ray tube housing assembly need not be hand-held during exposures.
In addition to the applicable provisions of 3.0, 4.0, 6.0, the requirements of 7.0 apply to x-ray equipment and associated facilities used for dental intraoral radiography. Requirements for extraoral dental radiographic systems are covered in 6.0.
7.1 Radiation Exposure Control. Means shall be provided to initiate the radiation exposure by a deliberate action on the part of the operator, such as the depression of a switch. Radiation exposure shall not be initiated without such an action.
7.1.1 Exposure Control Location and Operator Protection.
7.2 Stationary x‑ray systems shall be required to have the x‑ray exposure control permanently mounted in a protected area, so that the operator is required to remain in that protected area during the entire exposure; and
7.3 Mobile and portable x‑ray systems which are:
7.3.1 Used for greater than one week in the same location, i.e., a room or suite, shall meet the requirements of 7.1;
7.3.2 Used for less than one week in the same location shall be provided with either a protective barrier at least 2 meters (6.5 feet) high for operator protection, or means to allow the operator to be at least 2.7 meters (9 feet) from the tube housing assembly while making exposures.
7.4 kVp Limitations. Dental x-ray machines with a nominal fixed kVp of less than 50 kVp shall not be used to make diagnostic dental radiographs of humans.
7.5 Administrative Controls.
7.5.1 Patient and film holding devices shall be used when the techniques permit.
7.5.2 The tube housing and the PID for a permanently mounted intraoral dental system shall not be hand-held during an exposure. Appendix B specifies requirements for the use of intraoral dental radiographic units designed to be hand-held during patient examination.
7.5.3 Dental fluoroscopy without image intensification shall not be used.
8.1 Definitions. In addition to the definitions provided in A.2 and 2.0 of these regulations, the following definitions shall be applicable to 8.0
"Computed tomography dose index" (CTDI) means the integral of the dose profile along a line perpendicular to the tomographic plane divided by the product of the nominal tomographic section thickness and the number of tomograms produced in a single scan, that is:
z = Position along a line perpendicular to the tomographic plane;
D(z) = Dose at position z;
T = Nominal tomographic section thickness;
n = Number of tomograms produced in a single scan.
This definition assumes that the dose profile is centered around z=0 and that, for a multiple tomogram system, the scan increment between adjacent scans is nT.
"Contrast scale" means the change in the linear attenuation coefficient per CTN relative to water, that is:
where:
 = Linear attenuation coefficient of the material of interest;
= Linear attenuation coefficient of the material of interest;
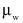 = Linear attenuation coefficient of water;
= Linear attenuation coefficient of water;
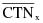 = of the material of interest;
= of the material of interest;
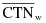 = of water.
= of water.
"CT conditions of operation" means all selectable parameters governing the operation of a CT x-ray system including nominal tomographic section thickness, filtration, and the technique factors as defined in 2.0.
"CT dosimetry phantom" means the phantom used for determination of the dose delivered by a CT x-ray system. The phantom shall be a right circular cylinder of polymethl-methacrylate of density 1.19±0.01 grams per cubic centimeter. The phantom shall be at least 14 centimeters in length and shall have diameters of 32.0 centimeters for testing any CT system designed to image any section of the body (whole body scanners) and 16.0 centimeters for any system designed to image the head (head scanners) or for any whole body scanner operated in the head scanning mode. The phantom shall provide means for the placement of a dosimeter(s) along its axis of rotation and along a line parallel to the axis of rotation 1.0 centimeter from the outer surface and within the phantom. Means for the placement of a dosimeter(s) or alignment device at other locations may be provided for convenience. Any effect on the doses measured due to the removal of phantom material to accommodate dosimeters shall be accounted for through appropriate corrections to the reported data or included in the statement of maximum deviation for the values obtained using the phantom.
"CT Number" means the number used to represent the x-ray attenuation associated with each elemental area of the CT image:
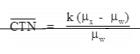
where:
k = A constant, a normal value of 1,000 when the Houndsfield scale of CTN is used;
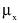 = Linear attenuation coefficient of the material of interest;
= Linear attenuation coefficient of the material of interest;
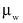 = Linear attenuation coefficient of water.
= Linear attenuation coefficient of water.
"Dose profile" means the dose as a function of position along a line.
"Modulation transfer function" means the modulus of the Fourier transform of the impulse response of the system.
"Multiple tomogram system" means a computed tomography x-ray system which obtains x-ray transmission data simultaneously during a single scan to produce more than one tomogram.
"Noise" means the standard deviation of the fluctuations in CTN expressed as a percentage of the attenuation coefficient of water. Its estimate (Sn) is calculated using the following expression:
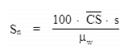
where:
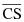 = Linear attenuation coefficient of the material of interest.
= Linear attenuation coefficient of the material of interest.
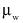 = Linear attenuation coefficient of water.
= Linear attenuation coefficient of water.
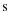 = Standard deviation of the CTN of picture elements in a specified area of the CT image.
= Standard deviation of the CTN of picture elements in a specified area of the CT image.
"Nominal tomographic section thickness" means the full width at half-maximum of the sensitivity profile taken at the center of the cross-sectional volume over which x-ray transmission data are collected.
"Picture element" means an elemental area of a tomogram.
"Reference plane" means a plane which is displaced from and parallel to the tomographic plane.
"Remanufacturing" means modifying a CT system in such a way that the resulting dose and imaging performance become substantially equivalent to any CT x-ray system manufactured by the original manufacturer on or after November 29, 1984. Any reference in this subsection to “manufacture,” “manufacturer,” or “manufacturing” includes remanufacture, remanufacturing, respectively.
"Scan" means the complete process of collecting x-ray transmission data for the production of a tomogram. Data can be collected simultaneously during a single scan for the production of one or more tomograms.
"Scan increment" means the amount of relative displacement of the patient with respect to the CT x-ray system between successive scans measured along the direction of such displacement.
"Scan sequence" means a pre-selected set of two or more scans performed consecutively under pre-selected CT conditions of operation.
"Sensitivity profile" means the relative response of the CT x-ray system as a function of position along a line perpendicular to the tomographic plane.
"Single tomogram system" means a CT x-ray system which obtains x-ray transmission data during a scan to produce a single tomogram.
"Tomographic plane" means that geometric plane which the manufacturer identified as corresponding to the output tomogram.
"Tomographic section" means the volume of an object whose x-ray attenuation properties are imaged in a tomogram.
8.2 Requirements for Equipment.
8.2.1 Termination of Exposure.
8.2.1.1 Means shall be provided to terminate the x-ray exposure automatically by either de-energizing the x-ray source or shuttering the x-ray beam in the event of equipment failure affecting data collection. Such termination shall occur within an interval that limits the total scan time to no more than 110 percent of its preset value through the use of either a backup timer or devices which monitor equipment function.
8.2.1.2 A visible signal shall indicate when the x-ray exposure has been terminated through the means required by Subdivision 8.2.1.1.
8.2.1.3 The operator shall be able to terminate the x-ray exposure at any time during a scan, or series of scans under CT x-ray system control, of greater than one-half second duration.
8.3 Tomographic Plane Indication and Alignment.
8.3.1 For any single tomogram system, means shall be provided to permit visual determination of the tomographic plane or a reference plane offset from the tomographic plane.
8.3.2 For any multiple tomogram system, means shall be provided to permit visual determination of the location of a reference plane. This reference plane can be offset from the location of the tomographic planes.
8.3.3 If a device using a light source is used to satisfy the requirements of Subdivisions 8.3.1 or 8.3.2, the light source shall provide illumination levels sufficient to permit visual determination of the location of the tomographic plane or reference plane under ambient light conditions of up to 500 lux.
8.4 Beam-On and Shutter Status Indicators and Control Switches.
8.4.1 The CT x-ray control and gantry shall provide visual indication whenever x-rays are produced and, if applicable, whether the shutter is open or closed.
8.4.2 Each emergency button or switch shall be clearly labeled as to its function.
8.5 Indication of CT Conditions of Operation. The CT x-ray system shall be designed such that the CT conditions of operation to be used during a scan or a scan sequence shall be indicated prior to the initiation of a scan or a scan sequence. On equipment having all or some of these conditions of operation at fixed values, this requirement may be met by permanent markings. Indication of CT conditions of operation shall be visible from any position from which scan initiation is possible.
8.6 Extraneous Radiation. When data are not being collected for image production, the radiation adjacent to the tube port shall not exceed that permitted by 4.3.
8.7 Maximum Surface CTDI Identification. The angular position where the maximum surface CTDI occurs shall be identified to allow for reproducible positioning of a CT dosimetry phantom.
8.8 Additional Requirements Applicable to CT X-Ray Systems Containing a Gantry Manufactured After September 3, 1985.
8.8.1 The total error in the indicated location of the tomographic plane or reference plane shall not exceed 5 millimeters.
8.8.2 If the x-ray production period is less than one-half second, the indication of x-ray production shall be actuated for at least one-half second. Indicators at or near the gantry shall be discernible from any point external to the patient opening where insertion of any part of the human body into the primary beam is possible.
8.8.3 The deviation of indicated scan increment versus actual increment shall not exceed plus or minus 1 millimeter with any mass from 0 to 100 kilograms resting on the support device. The patient support device shall be incremented from a typical starting position to the maximum incremented distance or 30 centimeters, whichever is less, and then returned to the starting position. Measurement of actual versus indicated scan increment may be taken anywhere along this travel.
8.8.4 Premature termination of the x-ray exposure by the operator shall necessitate resetting of the CT conditions of operation prior to the initiation of another scan.
8.9 Facility Design Requirements.
8.9.1 Aural Communication. Provision shall be made for two-way aural communication between the patient and the operator at the control panel.
8.9.2 Viewing Systems.
8.9.2.1 Windows, mirrors, closed-circuit television, or an equivalent shall be provided to permit continuous observation of the patient during irradiation and shall be so located that the operator can observe the patient from the control panel.
8.9.2.2 When the primary viewing system is by electronic means, an alternate viewing system (which may be electronic) shall be available for use in the event of failure of the primary viewing system.
8.10 Surveys, Calibrations, Spot Checks, and Operating Procedures.
8.11 Surveys.
8.11.1 All CT x-ray systems installed after [insert the effective date of the regulations] and those systems not previously surveyed shall have a survey made by, or under the direction of, a qualified medical physicist. In addition, such surveys shall be done after any change in the facility or equipment which might cause a significant increase in radiation hazard.
8.11.2 The registrant [licensee] shall obtain a written report of the survey from the qualified medical physicist, and a copy of the report shall be made available to the Agency upon request.
8.12 Radiation Calibrations.
8.12.1 The calibration of the radiation output of the CT x-ray system shall be performed by, or under the direction of, a qualified medical physicist who is physically present at the facility during such calibration.
8.12.2 The calibration of a CT x-ray system shall be performed after initial installation and before use on human patients, annually or at intervals specified by a qualified medical physicist, and after any change or replacement of components which, in the opinion of the qualified medical physicist, could cause a change in the radiation output.
8.12.3 The calibration of the radiation output of a CT x-ray system shall be performed with a calibrated dosimetry system. The calibration of such system shall be traceable to a national standard. The dosimetry system shall have been calibrated within the preceding 2 years.
8.12.4 CT dosimetry phantom(s) shall be used in determining the radiation output of a CT x-ray system. Such phantom(s) shall meet the following specifications and conditions of use:
8.12.4.1 CT dosimetry phantom(s) shall be right circular cylinders of polymethyl methacrylate of density 1.19 plus or minus 0.01 grams per cubic centimeter. The phantom(s) shall be at least 14 centimeters in length and shall have diameters of 32.0 centimeters for testing CT x-ray systems designed to image any section of the body and 16.0 centimeters for systems designed to image the head or for whole body scanners operated in the head scanning mode;
8.12.4.2 CT dosimetry phantom(s) shall provide means for the placement of a dosimeter(s) along the axis of rotation and along a line parallel to the axis of rotation 1.0 centimeter from the outer surface and within the phantom. Means for the placement of dosimeters or alignment devices at other locations may be provided;
8.12.4.3 Any effects on the doses measured due to the removal of phantom material to accommodate dosimeters shall be accounted for through appropriate corrections to the reported data or included in the statement of maximum deviation for the values obtained using the phantom;
8.12.4.4 All dose measurements shall be performed with the CT dosimetry phantom placed on the patient couch or support device without additional attenuation materials present.
8.12.5 The calibration shall be required for each type of head, body, or whole-body scan performed at the facility.
8.12.6 Calibration shall meet the following requirements:
8.12.6.1 The dose profile along the center axis of the CT dosimetry phantom for the minimum, maximum, and midrange values of the nominal tomographic section thickness used by the registrant shall be measurable. Where less than 3 nominal tomographic thicknesses can be selected, the dose profile determination shall be performed for each available nominal tomographic section thickness;
8.12.6.2 The CTDI2/ along the two axes specified in Subdivision 8.12.4.2 shall be measured. The CT dosimetry phantom shall be oriented so that the measurement point 1.0 centimeter from the outer surface and within the phantom is in the same angular position within the gantry as the point of maximum surface CTDI identified. The CT conditions of operation shall correspond to typical values used by the registrant;
8.12.6.3 The spot checks specified in 8.13 shall be made.
8.12.7 Calibration procedures shall be in writing. Records of calibrations performed shall be maintained for inspection by the Agency.
8.13 Spot Checks.
8.13.1 The spot-check procedures shall be in writing and shall have been developed by a qualified medical physicist.
8.13.2 The spot-check procedures shall incorporate the use of a CT dosimetry phantom which has a capability of providing an indication of contrast scale, noise, nominal tomographic section thickness, the resolution capability of the system for low and high contrast objects, and measuring the mean CTN for water or other reference material.
8.13.3 All spot checks shall be included in the calibration required by 8.11.2. and at time intervals and under system conditions specified by a qualified medical physicist.
8.13.4 Spot checks shall include acquisition of images obtained with the CT dosimetry phantom(s) using the same processing mode and CT conditions of operation as are used to perform calibrations required by 8.11.2. The images shall be retained, until a new calibration is performed, in two forms as follows:
8.13.4.1 Photographic copies of the images obtained from the image display device; and
8.13.4.2 Images stored in digital form on a storage medium compatible with the CT x-ray system.
8.13.5 Written records of the spot checks performed shall be maintained for inspection by the Agency.
8.14 Operating Procedures.
8.14.1 The CT x-ray system shall not be operated except by an individual who has been specifically trained in its operation.
8.14.2 Information shall be available at the control panel regarding the operation and calibration of the system. Such information shall include the following:
8.14.2.1 Dates of the latest calibration and spot checks and the location within the facility where the results of those tests may be obtained;
8.14.2.2 Instructions on the use of the CT dosimetry phantom(s) including a schedule of spot checks appropriate for the system, allowable variations for the indicated parameters, and the results of at least the most recent spot checks conducted on the system;
8.14.2.3 The distance in millimeters between the tomographic plane and the reference plane if a reference plane is utilized; and
8.14.2.4 A current technique chart available at the control panel which specifies for each routine examination the CT conditions of operation and the number of scans per examination.
8.14.3 If the calibration or spot check of the CT x-ray system identifies that a system operating parameter has exceeded a tolerance established by the qualified medical physicist, use of the CT x-ray system on patients shall be limited to those uses permitted by established written instructions of the qualified medical physicist.
9.1 Requirements for Certification.
9.1.1 Only x-ray systems, pursuant to the Mammography Quality Standards Reauthorization Act of 1998, Public Law 105-248, and 21 C.F.R. Part 900, shall be used for screening and diagnostic mammography.
9.1.2 A facility performing mammography shall have a valid certificate issued by the U.S. Department of Health and Human Services, pursuant to the Mammography Quality Standards Reauthorization Act of 1998, Public Law 105-248, and 21 C.F.R. Part 900.
9.1.3 A facility performing mammography shall ensure that the additional mammography activities of processing the x-ray film, interpreting the image, and maintaining viewing conditions, wherever performed, meet all quality standards pursuant to the Mammography Quality Standards Reauthorization Act of 1998, Public Law 105-248, and 21 C.F.R. Part 900.
[Current] federal regulations, 21 CFR Part 900, [dated] referring to states with certifying authority, is incorporated in its entirety in Part F.
11.1 Bone densitometry systems shall be:
11.1.1 Certified by the manufacturer pursuant to the Medical Device Act and Subchapter C – Electronic Product Radiation Control (EPRC) of Chapter V of the Federal Food, Drug and Cosmetic Act;
11.1.2 Registered [licensed] in accordance with Part B of these regulations; and
11.1.3 Maintained and operated in accordance with the manufacturer’s specifications.
11.2 Equipment Requirements. Systems with stepless collimators shall be provided with means to both size and align the x-ray field such that the x-ray field at the plane of the image receptor does not extend beyond 2 percent of the SID.
11.3 Operators of bone densitometry systems shall be:
11.3.1 Licensed, certified, or permitted as a radiologic technologist or technician [by the Agency]; or
11.3.2 Licensed as a practitioner of the healing arts; or
11.3.3 Permitted, certified, or approved [by the Agency] as a bone densitometry operator
11.4 During the operation of any bone densitometry system:
11.4.1 The operator, ancillary personnel, and members of the general public shall be positioned at least one meter from the patient and bone densitometry system during the examination.
11.5 The registrant [licensee] shall keep maintenance records for bone densitometry systems as prescribed by 11.1.3. These records shall be maintained for inspection by the Agency.
11.6 Bone densitometry on human patients shall be conducted only:
11.6.1 Under a prescription of a licensed practitioner of the healing arts; or
11.6.2 Under a screening program approved by the Agency.
11.7 Any person proposing to conduct a bone densitometry screening program shall submit the information outlined in Appendix A of this Part with the exception of g., h., i., j., k., and m., and include the name and address of the individual who will interpret the screening results.
12.1 All registrants [licensees] of diagnostic x-ray imaging equipment shall establish and maintain a quality assurance program consisting of quality control assessments addressing at least the following items:
12.1.1 Administration.
12.1.1.1 Written standard operating procedures on radiation protection and the practice of radiologic technology reviewed and updated annually by management;
12.1.1.2 Employee review and written acknowledgment of standard operating procedures and policies on radiation protection and the practice of radiologic technology;
12.1.1.3 Credentialing of practitioners, medical physicists, and x-ray equipment operators; and
12.1.1.4 Record retention in accordance with state statutes, regulations, but in no case less than three years.
12.1.2 Film Processing equipment.
12.1.2.1 Compliance with Section 3.0;
12.1.2.2 Film processor performance to include medium density, density difference, and base + fog;
12.1.2.3 Darkroom fog;
12.1.3 Radiographic equipment.
12.1.3.1 Compliance with performance standards in Sections 4.0 and 6.0;
12.1.3.2 Entrance skin exposure rates of selected patient examinations;
12.1.3.3 Image printing and viewing equipment;
12.1.3.4 Measurement of low and high contrast resolution; and
12.1.3.5 Radiation protection.
12.1.4 Fluoroscopic equipment.
12.1.4.1 Compliance with performance standards in Sections 4.0 and 5.0;
12.1.4.2 Entrance skin exposure rates of selected patient examinations;
12.1.4.3 Image printing and viewing equipment;
12.1.4.4 Measurement of low and high contrast resolution; and
12.1.4.5 Radiation protection.
12.1.5 Computerized tomography equipment.
12.1.5.1 Compliance with performance standards in Section 8.0;
12.1.5.2 CT number;
12.1.5.3 Low contrast and high contrast resolution;
12.1.5.4 Dosimetry of selected patient examinations to include pediatric patients if applicable;
12.1.5.5 Image printing and viewing equipment; and
12.1.5.6 Radiation protection.
12.1.6 Bone densitometry equipment.
12.1.6.1 Compliance with requirements in Section 11.0
12.1.7 Structural shielding for new facilities with x-ray equipment.
12.1.7.1 Pre-construction shielding design and evaluation; and
12.2 Post-construction radiation protection survey.
12.2.1 Structural shielding for modifying use or equipment in existing facility.
12.2.1.1 Re-evaluation of shielding design; and
12.2.1.2 Post-modification radiation protection survey.
12.3 The registrant [licensee] shall assign qualified personnel to fully implement the quality assurance program.
12.4 Quality control assessments for Section 12.1.1 may be assigned to qualified personnel who possess the requisite training and/or experience.
12.5 Quality control assessments for Section 12.1.1 shall be conducted by, or under the direction of, a qualified medical physicist.
12.6 The registrant [licensee] and/or qualified medical physicist shall determine the frequency of quality control tests.
12.7 The quality assurance program shall be in written form and available for review by the Agency.
12.8 Equipment used for compliance with the provisions of this section shall be properly calibrated and maintained in accordance with accepted professional standards.
This Section does not pertain to quality assurance for mammography equipment.
INFORMATION TO BE SUBMITTED BY PERSONS PROPOSING TO CONDUCT HEALING ARTS SCREENING
Persons requesting that the Agency approve a healing arts screening program shall submit the following information and evaluation:
1. Name and address of the applicant and, where applicable, the names and addresses of agents within this State;
2. Diseases or conditions for which the x‑ray examinations are to be used in diagnoses;
3. A description of the x‑ray examinations proposed in the screening program i.e., type and number of views;
4. Description of the population to be examined in the screening program, i.e., age range, sex, physical condition, and other appropriate information;
5. An evaluation of any known alternate methods not involving ionizing radiation that could achieve the goals of the screening program and why these methods are not used instead of the x‑ray examinations;
6. An evaluation by a qualified medical physicist of the x‑ray system(s) to be used in the screening program. The evaluation shall include the following:
a. Documentation that such system(s) satisfy all requirements of these regulations;
b. measurement of patient exposures from the x-ray examinations to be performed;
7. A description of the diagnostic x-ray quality control program;]
8. A copy of the technique chart for the x‑ray examination procedures to be used;
9. The qualifications of each individual who will be operating the x‑ray system(s);
10. The qualifications of the individual who will be supervising the operators of the x‑ray system(s). The extent of supervision and the method of work performance evaluation shall be specified;
11. The name and address of the practitioner licensed in the state who will interpret the radiograph(s);
12. Procedures to be used in advising the individuals screened and their practitioner of the healing arts or health care provider of the results of the screening procedure and any further medical needs indicated;
13. Procedures for the retention or disposition of the radiographs and other records pertaining to the x‑ray examinations;
14. Frequency of screening of individuals; and
15. The duration of the screening program.
HAND-HELD INTRAORAL DENTAL RADIOGRAPHIC UNIT REQUIREMENTS FOR USE
This appendix list the required information/documentation one must provide when requesting a variance for the use of a hand held unit. Such variance shall be specific to the particular facility location only.
The use of hand-held intraoral dental radiographic units must be approved as a variance by the Authority on Radiation Protection Board. Applications for variance must specify the conditions of use and precautions that will be put in place to assure safety and health protection equivalent to that of a stationary device.
The Agency may specify any of, but not be limited to, the following requirements for intraoral dental radiographic units designed to be operated as a hand-held unit:
1. For all uses:
a. The use of hand held intraoral dental radiographic units must be approved by the agency prior to use.
b. Operators of hand-held intraoral dental radiographic units shall be specifically trained to operate such equipment.
c. When operating a hand-held intraoral dental radiographic unit, operators shall wear a lead apron and thyroid collar, unless otherwise authorized by the Agency or a qualified health or medical physicist.
d. A hand-held intraoral dental radiographic unit shall be held without any motion during a patient examination. A tube stand may be utilized to immobilize a hand-held intraoral dental radiographic unit during patient examination.
e. Unless otherwise authorized by the Agency, a hand-held intraoral dental radiographic unit shall be used with a secondary radiation block.
f. The operator shall ensure there are no bystanders within a radius of at least six feet from the patient being examined with a hand-held intraoral radiographic unit.
2. Additional requirements for operatories in permanent facilities:
a. Hand-held intraoral dental radiographic units shall be used for patient examinations in dental operatories that meet the structural shielding requirements specified by the Agency or by a qualified health or medical physicist.
b. Hand-held intraoral dental radiographic units shall not be used for patient examinations in hallways and waiting rooms.
3. The following sections prohibit the use of hand held devices:
a. 3.1.9.3. Portable or mobile x-ray equipment shall be used only for examinations where it is impractical to transfer the patient(s) to a stationary x-ray installation. The use of hand held devices must be approved by the Agency utilizing the criteria in Appendix B.
b. 6.28.3.2. Mobile and Portable Systems. Mobile and portable x-ray systems which are:
6.28.3.2.1 Used continuously for greater than one week in the same location, i.e., a room or suite, shall meet the requirements of 6.28.3.1;
6.28.3.2.2 Used for less than one week at the same location shall be provided with either a protective barrier at least 2 meters (6.5 feet) high for operator protection during exposures, or means shall be provided to allow the operator to be at least 2.7 meters (9 feet) from the tube housing assembly during the exposure.
c. 6.30 Tube Stands for Portable X-Ray Systems. A tube stand or other mechanical support shall be used for portable x-ray systems, so that the x-ray tube housing assembly need not be hand-held during exposures.
d. 7.5.2 The tube housing and the PID for a permanently mounted intraoral dental system shall not be hand-held during an exposure. Appendix B specifies requirements for the use of intraoral dental radiographic units designed to be hand-held during patient examination.
MEDICAL DIAGNOSTIC AND INTERVENTIONAL X-RAY AND IMAGING SYSTEMS
This Part establishes requirements, for which a registrant is responsible, for use of diagnostic and interventional x-ray equipment and imaging systems by, or under the supervision of, an individual authorized by and licensed in accordance with State statutes to engage in the healing arts (dental, medical, or veterinary medicine). The provisions of this Part are in addition to, and not in substitution for, other applicable provisions of Parts A, B, D, G J, I, and X, of these regulations.
As used in this Part, the following definitions apply:
"Accessible surface" means the external surface of the enclosure or housing of the radiation producing machine as provided by the manufacturer.
“Agency” means the Division of Public Health, Delaware Department of Health and Social Services.
"Air kerma" means kerma in air (see definition of Kerma).
"Air kerma rate (AKR)" means the air kerma per unit time.
"Alert value" means a dose index (e.g., of CTDIvol (mGy) or DLP (mGy-cm)) that is set by the registrant to trigger an alert to the CT operator prior to scanning within an ongoing examination. The Alert value represents a universal dose index value well above the registrant established range for the examination that warrants more stringent review and consideration before proceeding.
"Aluminum equivalent" means the thickness of type 1100 aluminum alloy / affording the same attenuation, under specified conditions, as the material in question.
“Annual” means approximately every 12 months and not to exceed 14 months.
"Articulated joint" means a joint between two separate sections of a tabletop which joint provides the capacity of one of the sections to pivot on the line segment along which the sections join.
"Attenuation block" means a block or stack of type 1100 aluminum alloy, or aluminum alloy having equivalent attenuation, with dimensions 20 centimeters (cm) or larger by 20 cm or larger by 3.8 cm, that is large enough to intercept the entire x-ray beam.
"Automatic exposure control (AEC)" means a device which automatically controls one or more technique factors in order to obtain at a preselected location(s) a required quantity of radiation.
"Automatic exposure rate control (AERC)" means a device which automatically controls one or more technique factors in order to obtain, at a preselected location(s), a required quantity of radiation per unit time.
"Barrier" (See "Protective barrier").
"Beam axis" means a line from the source through the centers of the x-ray fields.
"Beam-limiting device" means a device which provides a means to restrict the dimensions of the x- ray field.
"Bone densitometer" means a device intended for medical purposes to measure bone density and mineral content by x-ray or gamma ray transmission measurements through the bone and adjacent tissues. This generic type of device may include signal analysis and display equipment, patient and equipment supports, component parts, and accessories.
"Bone densitometry" means a noninvasive measurement of certain physical characteristics of bone that reflect bone strength. Test results are typically reported as bone mineral content or density and are used for diagnosing osteoporosis, estimating fracture risk, and monitoring changes in bone mineral content.
"C-arm fluoroscope" means a fluoroscopic x-ray system in which the image receptor and the x-ray tube housing assembly are connected or coordinated to maintain a spatial relationship. Such a system allows a change in the direction of the beam axis with respect to the patient without moving the patient.
"Cantilevered tabletop" means a tabletop designed such that the unsupported portion can be extended at least 100 cm beyond the support.
"Cassette holder" means a device, other than a spot-film device, that supports and/or fixes the position of the image receptor during a radiographic exposure.
"Coefficient of variation (C)" means the ratio of the standard deviation to the mean value of a population of observations. It is estimated using the following equation:
where:
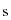 = Estimated standard deviation of the population.
= Estimated standard deviation of the population.
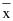 = Mean value of observations in sample;
= Mean value of observations in sample;
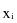 = ith observation in sample;
= ith observation in sample;
n = Number of observations sampled.
"Computed radiography (CR; also see DR)" means a digital x-ray imaging method in which a photo-stimulable phosphor is used to capture and store a latent image. The latent image is read out by stimulating the phosphor with a laser. Computed radiography systems may use cassettes to house the phosphor, or it may be integrated into a digital radiography system.
"Computed tomography (CT)" means the production of a tomogram by the acquisition and computer processing of x-ray transmission data.
"Computed tomography dose index" (CTDI) means the average absorbed dose, along the z-axis, from a series of contiguous irradiations. It is measured from one axial CT scan (one rotation of the x-ray tube), and is calculated by dividing the integrated absorbed dose by the nominal total beam collimation. The scattering media for CTDI consist of two (16 and 32 cm in diameter) polymethylmethacrylate (PMMA, e.g., acrylic or Lucite) cylinders of 14 cm length. The equation is: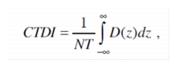
Where: D(z) = the radiation dose profile along the z-axis,
N = the number of tomographic sections imaged in a single axial scan. This is equal to the number of data channels used in a particular scan. The value of N may be less than or equal to the maximum number of data channels available on the system, and
T = the width of the tomographic section along the z-axis imaged by one data channel. In multiple-detector-row (multislice) CT scanners, several detector elements may be grouped together to form one data channel. In single-detector-row (single-slice) CT, the z-axis collimation (T) is the nominal scan width.
"Cone Beam Computed Tomography (CBCT)" is a volumetric imaging modality. Volumetric data are acquired using two dimensional digital detector arrays, and a cone-shaped x-ray beam (instead of fan-shaped) that rotates around the patient. Reconstruction algorithms can be used to generate images of any desired plane.
“Cone Beam CT” (CBCT) means a method of computed tomography that creates a three-dimensional cone-shaped beam that circles the patient once to generate an image.
"Control panel" means that part of the x-ray control upon which are mounted the switches, knobs, pushbuttons, keypads, touchscreens, and other hardware necessary for manually setting the technique factors.
"Cradle" means:
(1) A removable device which supports and may restrain a patient above an x-ray table; or
(2) A device;
(i) Whose patient support structure is interposed between the patient and the image receptor during normal use;
(ii) Which is equipped with means for patient restraint; and
(iii) Which is capable of rotation about its long (longitudinal) axis.
"CT" (See "Computed tomography").
"CT conditions of operation" means all selectable parameters governing the operation of a CT x-ray system including nominal tomographic section thickness, filtration, and the technique factors as defined in Part F, Section 2.0.
"CTDI100" means the accumulated multiple scan dose at the center of a 100-mm scan and underestimates the accumulated dose for longer scan lengths. It is thus smaller than the equilibrium dose. The CTDI100 requires integration of the radiation dose profile from a single axial scan over specific integration limits. In the case of CTDI100, the integration limits are +50 mm, which corresponds to the 100-mm length of the commercially available “pencil” ionization chamber. CTDI100 is acquired using a 100-mm long, 3-cc active volume CT “pencil” ionization chamber and one of the two standard CTDI acrylic phantoms (16 and 32 cm diameter) and a stationary patient table. The equation is:
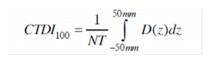
"CTDIvol" see "Volume Computed Tomography Dose Index (CTDIvol)"
"CTDIw" see "Weighted Computed Tomography Dose Index (CTDIw)"
"CT gantry" means tube housing assemblies, beam-limiting devices, detectors, and the supporting structures, frames, and covers which hold and/or enclose these components within a computed tomography system.
"CT number" means the number used to represent the x-ray attenuation associated with each elemental area of the CT image: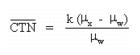
where:
k = A constant, a normal value of 1,000 when the Houndsfield scale of CT number is used;
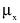 = Linear attenuation coefficient of the material of interest;
= Linear attenuation coefficient of the material of interest;
 = Linear attenuation coefficient of water.
= Linear attenuation coefficient of water.
"Cumulative air kerma" means the total air kerma accrued from the beginning of an examination or procedure and includes all contributions from fluoroscopic and radiographic irradiation.
"Detector" (See "Radiation detector")
"Diagnostic reference level" (DRL) is an investigational level used to identify unusually high radiation doses or dose rates for common medical X-ray imaging procedures. DRLs are suggested action levels above which a facility should review its methods and determine if acceptable image quality can be achieved at lower doses. DRLs should not be applied to an individual patient.
"Diagnostic source assembly" means the tube housing assembly with a beam-limiting device attached.
"Diagnostic x-ray system" means an x-ray system designed for irradiation of any part of the human [or animal] body for the purpose of diagnosis or visualization.
"Digital radiography (DR)" means an x-ray imaging method (or radiography) which produces a digital rather than analog image. DR includes both computed radiography and direct digital radiography.
"Direct digital radiography (DDR; also see CR and DR)" means an x-ray imaging method in which a digital sensor, usually incorporating a thin-film transistor, is used to capture an x-ray image. Some DDR systems use a scintillator to convert x-rays to light and a photodiode array to convert light to charge, while others use a photoconductor to convert x-rays directly to charge, which is stored on the thin-film transistor.
"Direct scattered radiation" means that scattered radiation which has been deviated in direction only by materials irradiated by the useful beam (See "Scattered radiation").
"Direct supervision" means a qualified, licensed practitioner must exercise general supervision, and be immediately available either physically or by an electronic device, to furnish assistance and direction throughout the performance of the procedures. It does not mean that the licensed practitioner must be present in the room when the procedure is being performed.
"Dose" means the absorbed dose as defined by the International Commission on Radiation Units and Measurements. The absorbed dose, D, is the quotient of de by dm, where de is the mean energy imparted to matter of mass dm; thus D=de/dm, in units of J/kg, where the special name of the unit of absorbed dose is gray (Gy).
"Dose area product (DAP) (aka kerma-area product (KAP))" means the product of the air kerma and the area of the irradiated field and is typically expressed in Gy-cm2, so it does not change with distance from the x-ray tube.
"Dose length product (DLP)" means the indicator of the integrated radiation dose from a complete CT examination. It addresses the total scan length by the formula:
DLP (mGy-cm) = CTDIvol (mGy) x scan length (cm)
"Dose profile" means the dose as a function of position along a line.
"Effective dose (E)" means the sum of the tissue-weighted equivalent doses for the radiosensitive tissues and organs of the body. It is given by the expression E = ?T (wT HT), in which HT is the equivalent dose in tissue or organ T and wT is the tissue weighting factor for tissue or organ T. The unit of E and HT is joule per kilogram (J·kg-1), with the special name sievert (Sv).
"Equipment" (See "X-ray equipment") means x-ray equipment.
"Exposure (X)" means the quotient of dQ by dm where dQ is the absolute value of the total charge of the ions of one sign produced in air when all the electrons and positrons liberated or created by photons in air of mass dm are completely stopped in air; thus X=dQ/dm, in units of C/kg. A second meaning of exposure is the process or condition during which the x-ray tube produces x-ray radiation.
"Field emission equipment" means equipment which uses an x-ray tube in which electron emission from the cathode is due solely to the action of an electric field.
"Filter" means material placed in the useful beam to preferentially absorb selected radiations.
"Fluoroscopically-Guided Interventional (FGI) Procedures" means an interventional diagnostic or therapeutic procedure performed via percutaneous or other access routes, usually with local anesthesia or intravenous sedation, which uses external ionizing radiation in the form of fluoroscopy to localize or characterize a lesion, diagnostic site, or treatment site, to monitor the procedure, and to control and document therapy.
"Fluoroscopic imaging assembly" means a subsystem in which x-ray photons produce a set of fluoroscopic images or radiographic images recorded from the fluoroscopic image receptor. It includes the image receptor(s), electrical interlocks, if any, and structural material providing linkage between the image receptor and diagnostic source assembly.
"Fluoroscopic irradiation time" means the cumulative duration during an examination or procedure of operator-applied continuous pressure to the device, enabling x-ray tube activation in any fluoroscopic mode of operation.
"Fluoroscopy" means a technique for generating x-ray images and presenting them simultaneously and continuously as visible images. This term has the same meaning as the term "radioscopy" in the standards of the International Electrotechnical Commission.
"Focal spot (actual)" means the area projected on the anode of the x-ray tube bombarded by the electrons accelerated from the cathode and from which the useful beam originates.
"General purpose radiographic x-ray system" means any radiographic x-ray system which, by design, is not limited to radiographic examination of specific anatomical regions.
"General supervision" means the procedure is performed under the overall direction and control of the qualified practitioner but the qualified practitioner is not required to be physically present during the performance of the procedure.
"Half-value layer (HVL)" means the thickness of specified material which attenuates the beam of radiation to an extent such that the AKR is reduced by one-half of its original value. In this definition, the contribution of all scattered radiation, other than any which might be present initially in the beam concerned, is deemed to be excluded.
"Hand-held x-ray equipment" means x-ray equipment that is designed to be hand-held during operation.
"Healing arts screening" means the testing of human beings using x-ray machines for the detection or evaluation of health indications when such tests are not specifically and individually ordered by a licensed practitioner of the healing arts legally authorized to prescribe such x-ray tests for the purpose of diagnosis or treatment.
"Heat unit" means a unit of energy equal to the product of the peak kilovoltage, milliamperes, and seconds, i.e., kVp x mA x second.
"HVL" (See "Half-value layer").
"Image intensifier" means a device, installed in its housing, which instantaneously converts an x-ray pattern into a corresponding light image of higher intensity.
"Image receptor" means any device, such as a fluorescent screen, radiographic film, x-ray image intensifier tube, solid-state detector, or gaseous detector which transforms incident x-ray photons either into a visible image or into another form which can be made into a visible image by further transformations. In those cases where means are provided to preselect a portion of the image receptor, the term “image receptor” shall mean the preselected portion of the device.
"Irradiation" means the exposure of matter to ionizing radiation.
"Isocenter" means the center of the smallest sphere through which the beam axis passes when the equipment moves through a full range of rotations about its common center.
"Kerma" means the quantity defined by the International Commission on Radiation Units and Measurements. The kerma, K, is the quotient of dEtr by dm, where dEtr is the sum of the initial kinetic energies of all the charged particles liberated by uncharged particles in a mass dm of material; thus K=dEtr/dm, in units of J/kg, where the special name for the unit of kerma is gray (Gy). When the material is air, the quantity is referred to as "air kerma."
"Kerma-area product (KAP)" (See "dose area product")
"Kilovolts peak" (See "Peak tube potential").
"kV" means kilovolts.
"kVp" (See "Peak tube potential").
"kWs" means kilowatt second.
"Last-image hold (LIH) radiograph" means an image obtained either by retaining one or more fluoroscopic images, which may be temporarily integrated, at the end of a fluoroscopic exposure or by initiating a separate and distinct radiographic exposure automatically and immediately in conjunction with termination of the fluoroscopic exposure.
"Lead equivalent" means the thickness of lead affording the same attenuation, under specified conditions, as the material in question.
"Leakage radiation" means radiation emanating from the diagnostic source assembly except for:
(1) The useful beam; and
(2) Radiation produced when the exposure switch or timer is not activated.
"Leakage technique factors" means the technique factors associated with the diagnostic source assembly which are used in measuring leakage radiation. They are defined as follows:
(1) For diagnostic source assemblies intended for capacitor energy storage equipment, the maximum-rated peak tube potential and the maximum-rated number of exposures in an hour for operation at the maximum-rated peak tube potential with the quantity of charge per exposure being 10 millicoulombs (or 10 mAs) or the minimum obtainable from the unit, whichever is larger;
(2) For diagnostic source assemblies intended for field emission equipment rated for pulsed operation, the maximum-rated peak tube potential and the maximum-rated number of x-ray pulses in an hour for operation at the maximum-rated peak tube potential; and
(3) For all other diagnostic source assemblies, the maximum-rated peak tube potential and the maximum-rated continuous tube current for the maximum-rated peak tube potential.
“Licensed practitioner" means an individual licensed to practice medicine, dentistry, podiatry, chiropractic, osteopathy, advanced practice nursing, or veterinary medicine in this state.
"Light field" means that area of the intersection of the light beam from the beam-limiting device and one of the set of planes parallel to and including the plane of the image receptor, whose perimeter is the locus of points at which the illumination is one-fourth of the maximum in the intersection.
"Line-voltage regulation" means the difference between the no-load and the load line potentials expressed as a percent of the load line potential; that is,
Percent line-voltage regulation = 100 (Vn-Vl)/Vl
where:
Vn = No-load line potential; and
Vl = Load line potential.
"mA" means milliampere.
"mAs" means milliampere second.
"Medical event" means one or more of the following criteria have occurred:
a. Event which results in an unintended dose greater than:
i. 0.5 Gy (50 rad) to any organ or tissue, or
ii. 0.05 Sv (5 rem) total effective dose equivalent for the procedure, or
b. Event involves wrong patient or wrong site for entire diagnostic exam (procedure/service) and
i. exceeds 0.5 Gy (50 rad) to an organ or tissue, or
ii. exceeds 0.05 Sv (5 rem) total effective dose equivalent for the procedure, or
c. Involves any equipment failure, personnel error, accident, mishap or other unusual occurrence with the administration of ionizing radiation that exceeds:
i. 0.5 Gy (50 rad) to an organ or tissue, or
ii. 0.05 Sv (5 rem) total effective dose equivalent for the procedure.
"Mobile x-ray equipment" (See "X-ray equipment").
"Mode of operation" means, for fluoroscopic systems, a distinct method of fluoroscopy or radiography provided by the manufacturer and selected with a set of several technique factors or other control settings uniquely associated with the mode. The set of distinct technique factors and control settings for the mode may be selected by the operation of a single control. Examples of distinct modes of operation include normal fluoroscopy (analog or digital), high-level control fluoroscopy, cineradiography (analog and digital), digital subtraction angiography, electronic radiography using the fluoroscopic image receptor, and photospot recording. In a specific mode of operation, certain system variables affecting kerma, AKR, or image quality, such as image magnification, x-ray field size, pulse rate, pulse duration, number of pulses, source-image receptor distance (SID), or optical aperture, may be adjustable or may vary; their variation per se does not comprise a mode of operation different from the one that has been selected.
"Multiple tomogram system" means a computed tomography x-ray system which obtains x-ray transmission data simultaneously during a single scan to produce more than one tomogram.
"Noise" in CT means the standard deviation of the fluctuations in CT number expressed as a percentage of the attenuation coefficient of water. Its estimate (Sn) is calculated using the following expression: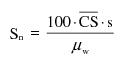
where:
 = Linear attenuation coefficient of the material of interest.
= Linear attenuation coefficient of the material of interest.
 = Linear attenuation coefficient of water.
= Linear attenuation coefficient of water.
 = Estimated [S]standard deviation of the CT numbers of picture elements in a specified area of the CT image.
= Estimated [S]standard deviation of the CT numbers of picture elements in a specified area of the CT image.
"Nominal tomographic section thickness" means the full width at half-maximum of the sensitivity profile taken at the center of the cross-sectional volume over which x-ray transmission data are collected.
"Notification value" means a protocol-specific dose index (e.g. CTDIvol(mGy) or of DLP(mGy-cm)) that is set by the registrant to trigger a notification to the CT operator prior to scanning when the dose index exceeds the established range for the examination.
"Patient" means an individual or animal subjected to healing arts examination, diagnosis or treatment.
"PBL" See "Positive beam limitation"
"Peak tube potential" means the maximum value of the potential difference across the x-ray tube during an exposure.
"Personal supervision" means a qualified practitioner must exercise General Supervision and be present in the room or adjacent control area during the performance of the procedure.
"Phantom" means a volume of material behaving in a manner similar to tissue with respect to the attenuation and scattering of radiation. This requires that both the atomic number (Z) and the density of the material be similar to that of tissue.
"Photostimulable storage phosphor (PSP)" means a material used to capture and store radiographic images in computed radiography systems.
"Picture element" means an elemental area of a tomogram.
"PID" (See "Position indicating device").
"Pitch" means the table incrementation, in CT, per x-ray tube rotation, divided by the nominal x-ray beam width at isocenter.
"Portable x-ray equipment" (See "X-ray equipment").
"Position indicating device (PID)" means a device on dental x-ray equipment used to indicate the beam position and to establish a definite source-surface (skin) distance. It may or may not incorporate or serve as a beam-limiting device.
"Positive beam limitation" means the automatic or semi-automatic adjustment of an x-ray beam to the size of the selected image receptor, whereby exposures cannot be made without such adjustment.
"Primary protective barrier" means the material, excluding filters, placed in the useful beam to reduce the radiation exposure [beyond the patient and cassette holder] for protection purposes.
"Protective apron" means an apron made of radiation absorbing materials used to reduce radiation exposure.
"Protocol" means a collection of settings and parameters that fully describe an examination.
"Pulsed mode" means operation of the x-ray system such that the x-ray tube current is pulsed by the x-ray control to produce one or more exposure intervals of duration less than one-half second.
“Qualified expert” means an individual who has satisfactorily fulfilled the training and experience requirements consistent with achieving a level of competency sufficient to function effectively in the position for which State Radiation Service Provider registration is sought in accordance with Regulation 4465, Part B. Such individuals must demonstrate to the satisfaction of the Agency their qualifications, for example formal education or individuals certified in the appropriate field by the American Board of Radiology, or the American Board of Health Physics, or the American Board of Medical Physics, or those having equivalent qualifications. With reference to the calibration of radiation therapy equipment, an individual, in addition to the above qualifications, must be qualified in accordance with Regulation 4465, Part F and Regulation 4465, Part X of these regulations, as amended.
"Qualified medical physicist (QMP)" means an individual who meets each of the following credentials:
1. Has earned a master's and/or doctoral degree in physics, medical physics, biophysics, radiological physics, medical health physics, or equivalent disciplines from an accredited college or university; and
2. Has been granted certification in the specific subfield(s) of medical physics with its associated medical health physics aspects by an appropriate national certifying body and abides by the certifying body's requirements for continuing education; and/or
3. Is credentialed in accordance with Part X, subsection 3.4, as amended.
"Quality Assurance" means a program providing for verification by written procedures such as testing, auditing, and inspection to ensure that deficiencies, deviations, defective equipment, or unsafe practices, or a combination thereof, relating to the use, disposal, management, or manufacture of radiation devices are identified, promptly corrected, and reported to the appropriate regulatory authorities as required.
"Radiation detector" means a device which in the presence of radiation provides a signal or other indication suitable for use in measuring one or more quantities of incident radiation.
"Radiation Protocol Committee (RPC)" means the representative group of qualified individuals in a CT or FGI facility responsible for the ongoing review and management of CT or FGI protocols to ensure that exams being performed achieve the desired diagnostic image quality at the lowest radiation dose possible while properly exploiting the capabilities of the equipment being used.
"Radiation therapy simulation system" means a radiographic or fluoroscopic x ray system intended for localizing the volume to be exposed during radiation therapy and confirming the position and size of the therapeutic irradiation field.
"Radiograph" means an image receptor on which the image is created directly or indirectly by an x-ray pattern and results in a permanent record.
"Radiography" means a technique for generating and recording an x-ray pattern for the purpose of providing the user with an image(s) after termination of the exposure.
"Recording" means producing a retrievable form of an image resulting from x-ray photons.
"Reference plane" means a plane which parallel to and which can be offset (as specified in manufacturer information provided to users) from the location of the tomographic plane(s).
"Scan" means the complete process of collecting x-ray transmission data for the production of a tomogram. Data may be collected simultaneously during a single scan for the production of one or more tomograms.
"Scan increment" means the amount of relative displacement of the patient with respect to the CT x-ray system between successive scans measured along the direction of such displacement.
"Scan sequence" means a pre-selected set of two or more scans performed consecutively under pre-selected CT conditions of operation.
"Scan time" means the time elapsed during the accumulation of x-ray transmission data for a single scan.
"Scattered radiation" means radiation that, during passage through matter, has been deviated in direction (See "Direct scattered radiation").
"Sensitivity profile" means the relative response of the CT x-ray system as a function of position along a line perpendicular to the tomographic plane.
"Shutter" means a device attached to the tube housing assembly which can intercept the entire cross sectional area of the useful beam and which has a lead equivalency not less than that of the tube housing assembly.
"SID" (See "Source-image receptor distance").
"Single tomogram system" means a CT x-ray system which obtains x-ray transmission data during a scan to produce a single tomogram.
"Size-specific dose estimate (SSDE)" means a patient dose estimate which takes into consideration corrections based on the size of the patient, using linear dimensions measured on the patient or patient images.
"Source" means the focal spot of the x-ray tube.
"Source-image receptor distance" means the distance from the source to the center of the input surface of the image receptor.
"Source-skin distance (SSD)" means the distance from the source to the center of the entrant x-ray field in the plane tangent to the patient skin surface.
"Spot-film" / means a radiograph which is made during a fluoroscopic examination to permanently record conditions which exist during that fluoroscopic procedure.
"Spot-film device" means a device intended to transport and/or position a radiographic image receptor between the x-ray source and fluoroscopic image receptor. It includes a device intended to hold a cassette over the input end of the fluoroscopic image receptor for the purpose of producing a radiograph.
"Stationary x-ray equipment" (See "X-ray equipment").
"Stray radiation" means the sum of leakage and scattered radiation.
"Substantial radiation dose level" (SRDL) means an appropriately-selected dose used to trigger additional dose-management actions during a procedure and medical follow-up for a radiation level that might produce a clinically-relevant injury in an average patient.
"Technique factors" means the following conditions of operation:
(1) For capacitor energy storage equipment, peak tube potential in kilovolts (kV) and quantity of charge in milliampere-seconds (mAs);
(2) For field emission equipment rated for pulsed operation, peak tube potential in kV, and number of x-ray pulses;
(3) For CT equipment designed for pulsed operation, peak tube potential in kV, scan time in seconds, and either tube current in milliamperes (mA), x-ray pulse width in seconds, and the number of x-ray pulses per scan, or the product of tube current, x-ray pulse width, and the number of x-ray pulses in mAs;
(4) For CT equipment not designed for pulsed operation, peak tube potential in kV, and either tube current in mA and scan time in seconds, or the product of tube current and exposure time in mAs and the scan time when the scan time and exposure time are equivalent; and
(5) For all other equipment, peak tube potential in kV, and either tube current in mA and exposure time in seconds, or the product of tube current and exposure time in mAs.
"Tomogram" means the depiction of the x-ray attenuation properties of a section through the body.
"Tomographic plane" means that geometric plane which the manufacturer identified as corresponding to the output tomogram.
"Tomographic section" means the volume of an object whose x-ray attenuation properties are imaged in a tomogram.
"Tube" means an x-ray tube, unless otherwise specified.
"Tube housing assembly" means the tube housing with tube installed. It includes high-voltage and/or filament transformers and other appropriate elements when such are contained within the tube housing.
"Unintended" radiation dose in diagnostic or interventional x-ray means a patient radiation dose determined to have resulted from a human error or equipment malfunction during the procedure.
"Useful beam" means the radiation which passes through the tube housing port and the aperture of the beam limiting device when the exposure switch or timer is activated.
"Visible area" means that portion of the input surface of the image receptor over which incident x-ray photons are producing a visible image.
"Volume Computed Tomography Dose Index (CTDIvol)" means a radiation dose parameter derived from the CTDIw (weighted or average CTDI given across the field of view). The formula is:
CTDIvol = (N)(T)(CTDIw)/I, where
N = number of simultaneous axial scans per x-ray source rotation,
T = thickness of one axial scan (mm), and
I = table increment per axial scan (mm).
Thus,
CTDIvol = CTDIw / pitch
"Weighted Computed Tomography Dose Index (CTDIw)" means the estimated average CTDI100 across the field of view (FOV). The equation is: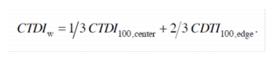
Where 1/3 and 2/3 approximate the relative areas represented by the center and edge values derived using the 16 or 32 cm acrylic phantom. CTDIw uses CTDI100 and an f-factor for air (0.87 rad/R or 1.0 mGy/mGy).
"X-ray control" means a device which controls input power to the x-ray high-voltage generator and/or the x-ray tube. It includes equipment such as timers, phototimers, automatic brightness stabilizers, and similar devices, which control the technique factors of an x-ray exposure.
"X-ray equipment" means an x-ray system, subsystem, or component thereof. Types of x-ray equipment are as follows:
(1) "Mobile x-ray equipment" means x-ray equipment mounted on a permanent base with wheels and/or casters for moving while completely assembled;
(2) "Portable x-ray equipment" means x-ray equipment designed to be hand-carried; and
(3) "Stationary x-ray equipment" means x-ray equipment which is installed in a fixed location.
(4) "Hand-held x-ray equipment" means x-ray equipment that is designed to be hand-held during operation.
"X-ray exposure control" means a device, switch, button or other similar means by which an operator initiates and/or terminates the radiation exposure. The x-ray exposure control may include such associated equipment as timers and back-up timers.
"X-ray field" means that area of the intersection of the useful beam and any one of a set of planes parallel to and including the plane of the image receptor, whose perimeter is the locus of points at which the AKR is one-fourth of the maximum in the intersection.
"X-ray high-voltage generator" means a device which transforms electrical energy from the potential supplied by the x-ray control to the tube operating potential. The device may also include means for trans-forming alternating current to direct current, filament transformers for the x-ray tube(s), high-voltage switches, electrical protective devices, and other appropriate elements.
"X-ray system" means an assemblage of components for the controlled production of x-rays. It includes minimally an x-ray high-voltage generator, an x-ray control, a tube housing assembly, a beam-limiting device, and the necessary supporting structures. Additional components which function with the system are considered integral parts of the system.
"X-ray table" means a patient support device with its patient support structure (tabletop) interposed between the patient and the image receptor during radiography and/or fluoroscopy. This includes, but is not limited to, any stretcher equipped with a radiolucent panel and any table equipped with a cassette tray (or bucky), cassette tunnel, fluoroscopic image receptor, or spot-film device beneath the tabletop.
"X-ray tube" means any electron tube which is designed for the conversion of electrical energy into x-ray energy.
3.1 Radiation Safety Requirements. The registrant shall be responsible for directing the operation of the x-ray system(s) under his or her administrative control and shall assure that the requirements of these regulations are met in the operation of the x-ray system(s).
3.1.1 The registrant shall have a radiation safety program. The radiation safety program shall include but not be limited to the following:
3.1.1.1 The use of ionizing radiation within its purview is performed in accordance with existing laws and regulations.
3.1.1.2 All persons are protected as required by Regulation 4465, Part D, Standards for Protection Against Radiation, of these regulations.
3.1.1.3 Upon discovery of a medical event, the registrant shall:
3.1.1.3.1 Contact the Agency regarding the medical event within one business day;
3.1.1.3.2 Provide a written report, including the analysis of the medical event, by a QMP (QE) to the Agency within 15 business days;
3.1.1.3.3 Provide a clinical summary to the prescribing physician and patient within 15 business days; and
3.1.1.4 Maintain record of the medical event as part of the patient's permanent medical record, and
3.1.1.5 Maintain record of the medical event as reported to Radiation Protocol or Safety Committee for at least three years, available for inspection.
3.1.2 An x-ray system which does not meet the provisions of these regulations shall not be operated for diagnostic or interventional purposes unless the Agency or a QMP (QE) determines that the non-compliance shall not pose a significant radiation risk or significantly affect image quality, and arrangements have been made to correct the non-compliance within 30 days.
3.1.3 The QMP (QE), if required in this Part, shall complete initial and routine compliance evaluations following nationally recognized procedures. These evaluations shall include a review of the required QC tests.
3.1.4 All x-ray equipment shall be installed, maintained, and used in accordance with the equipment manufacturer’s specifications.
3.1.5 Individuals operating the x-ray systems shall meet the qualifications required by the Agency.
3.1.6 A sufficient number of protective apparel (e.g., aprons, gloves, collars) and shields shall be available to provide the necessary radiation protection for all patients and personnel who are involved with x-ray operations.
3.1.7 All protective apparel and auxiliary shields shall be evaluated annually for integrity and clearly labeled with their lead equivalence.
3.1.8 Each registrant should have a mechanism in place for the referring licensed practitioner to access information on selecting the most appropriate diagnostic procedure to answer the clinical question.
3.1.9 Nationally recognized diagnostic reference levels (DRLs) should be utilized when applicable.
3.1.10 The registrant shall use dose reduction strategies designed to minimize patient and personnel exposure commensurate with the needed diagnostic information.
3.1.11 Portable or mobile x-ray equipment shall be used only for examinations where it is impractical to transfer the patient to a stationary x-ray installation.
3.1.12 Neither the x-ray tube housing nor the collimating device shall be held during an exposure. An exemption or variance s is required for devices specifically designed to be hand-held, except for devices in non-human use, such as veterinary medicine or forensics.
3.1.13 The useful x-ray beam shall be limited to the area of clinical interest.
3.1.14 Consideration shall be given to selecting the appropriate technique and employing available dose reduction methods and technologies across all patient sizes and clinical indications.
3.1.15 A facility shall have a documented procedure in place for verification of patient identity and exam to be performed, including identification of the appropriate body part.
3.1.16 For general radiographic systems not equipped with an operational anatomic programming option, protocols shall be documented and readily available to the operator. At a minimum, these protocols shall include:
3.1.16.1 Patient's (adult and pediatric, if appropriate) body part and anatomical size
3.1.16.2 Technique factors
3.1.16.3 Type of image receptor used
3.1.16.4 Source to image receptor distance used (except for dental intraoral radiography)
3.1.16.5 Type of grid, if any.
3.1.17 The registrant shall create and make available to x-ray operators written safety procedures, including instructions for patient holding and any restrictions of the operating technique required for the safe operation of the particular x-ray system. The operator shall be able to demonstrate familiarity with these procedures.
3.1.18 The registrant shall restrict the presence of individuals in the immediate area of the patient being examined to those required or in training for the medical procedure, or the parent or guardian of a patient while the x-ray tube is energized. The following applies to all individuals, other than the patient being examined:
3.1.18.1 All persons shall be positioned such that no part of the body will be struck by the useful beam unless protected by not less than 0.5 millimeter lead equivalent material;
3.1.18.2 All persons shall be protected from the secondary radiation by protective garments or whole body protective barriers of not less than 0.25 millimeter lead equivalent material;
3.1.18.3 Instances may warrant having human patients other than the one being examined in the room during the exam. If the procedure results in scatter radiation in excess of 0.02 mSv (2 mR) in any one hour at the position of these patients, they shall be protected from the direct scatter radiation by whole body protective barriers of not less than 0.25 millimeter lead equivalent material or shall be positioned so that the 0.02 mSv (2 mR) in any one hour limit is met.
3.1.19 Individuals shall not be exposed to the useful beam except for healing arts purposes and unless such exposure has been authorized by a licensed practitioner. This provision specifically prohibits deliberate exposure for the following purposes:
3.1.19.1 Exposure of an individual for training, demonstration, or other non-healing arts purposes; and
3.1.19.2 Exposure of an individual for the purpose of healing arts screening except as authorized by the Agency.
3.1.19.3 Except under the following circumstances:
3.1.19.3.1 Exposure of an individual to the useful beam when conducting research approved by an Institutional Review Board (IRB) as allowed by Title 45, Code of Federal Regulations (CFR), Part 46 and Title 21,CFR, Part 56.
3.1.20 In cases where a patient or image receptor must be provided with auxiliary support, mechanical support devices shall be used whenever possible. If a patient or image receptor must be provided with auxiliary support during a radiation exposure:
3.1.20.1 Written safety procedures, as required by Part F, subsection 3.1.15, shall indicate the requirements for selecting a holder and the procedure the holder shall follow;
3.1.20.2 The human holder shall be instructed in personal radiation safety and protected as required by Part F, subsection 3.1.16;
3.1.20.3 No individual shall be used routinely to hold the image receptor or patient during a radiation exposure;
3.1.20.4 In those cases where the patient must hold the image receptor, except during intraoral examinations, any portion of the body other than the area of clinical interest struck by the useful beam shall be protected by not less than 0.5 millimeter lead equivalent material.
3.1.21 All individuals who are associated with the operation of an x-ray system are subject to the requirements of Part D of these regulations.
3.1.22 Healing Arts Screening. Any person proposing to conduct a healing arts screening program shall not initiate such a program without prior approval of the Agency. When requesting such approval, that person shall submit the information outlined in Appendix A of Part F of these regulations. If any information submitted to the Agency becomes invalid or outdated, the Agency shall be immediately notified. FDA/MQSA-certified facilities are registered with the Agency for the use of dedicated mammographic equipment to conduct mammography screening.
3.1.23 Maintenance of Records. The registrant shall maintain the following information on each x-ray system for inspection by the Agency for a minimum of 5 years or as noted below:
3.1.23.1 Model and serial numbers of all major components, and user's manuals for those components, including software, shall be maintained for the life of the system.
3.1.23.2 Records of surveys, calibrations, maintenance, and modifications (e.g., major software and hardware upgrades) performed on the x-ray system(s); and
3.1.23.3 A copy of all correspondence with the Agency regarding the x-ray system.
3.1.24 X-Ray Utilization Record. Each facility shall maintain a record containing the patient's name, the type of examinations, and the dates the examinations were performed.
3.2 Quality Assurance.
3.2.1 The registrant shall establish and maintain a quality assurance (QA) program. In addition to the standards in the modality specific sections, the registrant shall:
3.2.1.1 Maintain documentation of minimum qualifications for practitioners, medical physicists, and x-ray equipment operators.
3.2.1.2 Designate an appropriately trained individual to manage the QA program.
3.2.1.3 Establish and maintain written QA and quality control (QC) procedures, including evaluation frequencies and tolerances.
3.2.1.4 Check each study for artifacts. If an artifact is present, the source shall be identified and appropriate action taken.
3.2.1.5 Perform repeat / reject analysis of radiographic images at least quarterly following specifications of a nationally recognized organization.
3.2.1.6 Complete preventative maintenance on the x-ray systems in accordance with manufacturer specifications at intervals not to exceed 12 months.
3.2.1.7 Maintain documentation showing the testing instruments used in determining compliance with the provisions of this section are properly calibrated and maintained in accordance with the Agency minimum standard or accepted professional standards when no Agency minimum is defined.
3.2.1.8 Complete and document an annual review of the QA program.
3.2.1.9 Retain QA/QC records of evaluations and reviews in accordance with state statutes, regulations, but in no case less than three years.
3.2.2 X-Ray Film Processing Facilities. A registrant using analog image receptors (e.g. radiographic film) shall have available suitable equipment for handling and processing radiographic film in accordance with the following provisions:
3.2.2.1 Manually developed film:
3.2.2.1.1 Processing tanks shall be constructed of mechanically rigid, corrosion resistant material; and
3.2.2.1.2 Developing solutions shall be prepared, replenished, and replaced following manufacturer recommendations.
3.2.2.1.3 The temperature of solutions in the tanks shall be maintained within the range of 60o F to 80o F (16o C to 27o C). Film shall be developed in accordance with the time-temperature relationships recommended by the film manufacturer, or, in the absence of such recommendations, with the following time-temperature chart: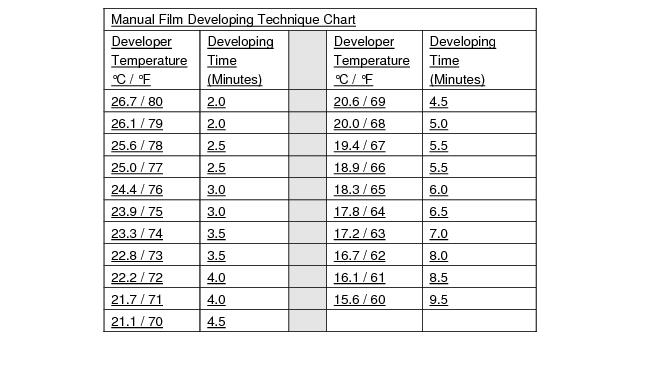
3.2.2.1.4 Devices shall be utilized which will indicate the actual temperature of the developer solution and signal the passage of a preset time.
3.2.3 Automatic processors and other closed processing systems:
3.2.3.1 Automatic processors shall be operated and maintained following manufacturer specifications.
3.2.3.2 Films shall be developed in accordance with the time-temperature relationships recommended by the film manufacturer; in the absence of such recommendations, the film shall be developed using the following chart:

3.2.3.3 Processing deviations from the requirements of Part F, subsection 3.2.2 shall be documented by the registrant in such manner that the requirements are shown to be met or exceeded (e.g., extended processing, and special rapid chemistry).
3.2.4 Additional Requirements for Facilities using X-ray Film.
3.2.4.1 Pass boxes, if provided, shall be so constructed as to exclude light from the darkroom when cassettes are placed in or removed from the boxes, and shall incorporate adequate shielding from stray radiation to prevent exposure of undeveloped film.
3.2.4.2 Darkrooms typically used by more than one individual shall be provided a method to prevent accidental entry while undeveloped films are being handled or processed.
3.2.4.3 Film shall be stored in a cool, dry place and shall be protected from exposure to stray radiation. Film in open packages shall be stored in a light tight container.
3.2.4.4 Film cassettes and intensifying screens shall be inspected periodically and shall be cleaned and replaced as necessary.
3.2.4.5 Outdated x-ray film shall not be used for diagnostic radiographs.
3.2.4.6 The film and intensifying screen shall be spectrally compatible.
3.2.4.7 Facilities shall maintain a light-tight darkroom, use proper safelighting and safeguards, and evaluate darkroom integrity and daylight loading systems for film fog every six months and after a change that may impact film fog.
3.2.4.8 Facilities other than dental, podiatry, and veterinary shall:
3.2.4.8.1 Have a continuous and documented sensitometric quality control program, including quality control tests for speed, contrast and fog, These tests shall be performed according to specifications of the manufacturer, a QMP (QE), or a nationally recognized organization.
3.2.4.8.2 Maintain a light-tight darkroom and use proper safelighting and safeguards such that any film type in use exposed in a cassette to x-radiation sufficient to produce an optical density from 1 to 2 when processed shall not suffer an increase in optical density greater than 0.1 when exposed in the darkroom for 2 minutes with all safelights on. If used, daylight film handling boxes shall preclude fogging of the film.
3.2.4.8.3 Limit the base plus fog of unexposed film to an optical density less than 0.25 when developed by the routine procedure used by the facility.
3.2.5 Facilities Using Computed Radiography (CR) or Direct Digital Radiography (DDR).
3.2.5.1 When exposure indicators are available, the facility shall establish and document an acceptable range for the exposure values for examinations routinely performed at the facility. The indicated exposure values for each image shall be compared to the established range. Consistent deviations from established ranges shall be investigated, corrective actions taken as necessary, and results documented.
3.2.5.2 Facilities shall establish and follow an image quality control program in accord with the recommendations of a QMP (QE), the system manufacturer, or a nationally recognized organization.
3.2.5.3 Facilities other than dental, podiatric and veterinary, shall quarterly complete phantom image evaluation using a phantom approved by a QMP (QE), system manufacturer, or the Agency. The analysis at a minimum shall include: artifacts, spatial resolution, contrast/noise, workstation monitors, and exposure indicator constancy.
3.2.5.4 In addition to Part F, subsection 3.2.4.1 through 3.2.4.3, CR facilities shall perform erasure of all CR cassettes, at least on a weekly basis.
3.3 Exemptions.
3.3.1 Dental facilities. Dental facilities performing only intra-oral, panoramic, cephalometric or volumetric dental imaging are exempt from the following provisions of this Section: Part F, subsection 3.1.8 (information available to referring physician) and Part F, subsection 3.2.1.5 (repeat analysis).
3.3.2 Podiatry facilities. Podiatry facilities are exempt from the following provisions of this Section: Part F, subsection 3.1.7 (information available to referring physician) and Part F, subsection 3.2.1.5 (repeat analysis).
3.3.3 Veterinary facilities. Veterinary facilities are exempt from the following provisions of this Section: Part F, subsection 3.1.8 (information available to referring physician), Part F, subsection 3.1.9 (use of reference levels), Part F, subsection 3.1.12 (use of dose reduction techniques), Part F, subsection 3.1.13 (patient identification), Part F, subsection 3.1.14 (protocol control), Part F, 3.1.18.3 (routine holding of patient), Part F, subsection 3.1.20 (healing arts screening), Part F, subsection 3.2.1.5 (repeat analysis), and Part F, subsection 3.2.3.8.1, (a) through (c) (use of sensitometric equipment).
4.1 In addition to other requirements of this Part, all diagnostic and interventional x ray systems shall meet the following requirements. Requirements specific to dental intra-oral, panoramic, cephalometric, volumetric dental imaging equipment are included in Part F, Section 7.0.
4.2 Warning Label.
4.2.1 On systems manufactured on or before June 10, 2006, the control panel containing the main power switch shall bear the warning statement, or the warning statement in Part F, subsection 4.2.2, legible and accessible to view: "WARNING: This x-ray unit may be dangerous to patient and operator unless safe exposure factors, operating instructions are observed."
4.2.2 On systems manufactured after June 10, 2006, the control panel containing the main power switch shall bear the warning statement, legible and accessible to view: "WARNING: This x-ray unit may be dangerous to patient and operator unless safe exposure factors, operating instructions and maintenance schedules are observed."
4.3 Leakage Radiation from the Diagnostic Source Assembly. The leakage radiation from the diagnostic source assembly measured at a distance of 1 meter in any direction from the source shall not exceed 0.88 milligray (mGy) air kerma (vice 100 milliroentgen (mR) exposure) in 1 hour when the x-ray tube is operated at its leakage technique factors. If the maximum rated peak tube potential of the tube housing assembly is greater than the maximum rated peak tube potential for the diagnostic source assembly, positive means shall be provided to limit the maximum x-ray tube potential to that of the diagnostic source assembly. Compliance shall be determined by measurements averaged over an area of 100 square centimeters with no linear dimension greater than 20 centimeters (21 CFR 1020.30(k)).
4.4 Radiation from Components Other Than the Diagnostic Source Assembly. The radiation emitted by a component other than the diagnostic source assembly shall not exceed an air kerma of 18 microgray (vice 2 milliroentgens exposure) in 1 hour at 5 centimeters from any accessible surface of the component when it is operated in an assembled x-ray system under any conditions for which it was designed. Compliance shall be determined by measurements averaged over an area of 100 square centimeters with no linear dimension greater than 20 centimeters. (21 CFR 1020.30(l))
4.5 Technique Indicators.
4.5.1 For x-ray equipment capable of displaying technique factors, the technique factors to be used during an exposure shall be indicated before the exposure begins. If automatic exposure controls are used, the technique factors which are set prior to the exposure shall be indicated. (21 CFR 1020.31(a)(1))
4.5.2 The requirement of Part F, subsection 4.5.1 may be met by permanent markings on equipment having fixed technique factors. Indication of technique factors shall be visible from the operator's position except in the case of spot films made by the fluoroscopist. (21 CFR 1020.31(a)(1))
4.5.3 The accuracy of the indicated kilovoltage peak (kVp) shall meet manufacturer specifications. In the absence of a manufacturer specification, kVp accuracy shall be within +10 percent.
4.6 Beam Quality.
4.6.1 The half value layer (HVL) of the useful beam for a given x-ray tube potential shall not be less than the values shown in Table 1. If it is necessary to determine such half-value layer at an x-ray tube potential which is not listed in Table 1 of this section, linear interpolation or extrapolation may be made. Positive means shall be provided to ensure that at least the minimum filtration needed to achieve beam quality requirements is in the useful beam during each exposure. (21 CFR 1020.30(m)) In the case of a system, which is to be operated with more than one thickness of filtration, this requirement can be met by a filter interlocked with the kilovoltage selector which will prevent x-ray emissions if the minimum required filtration is not in place. (21 CFR 1020.30)
TABLE 1
(21 CFR 1020.30(m))
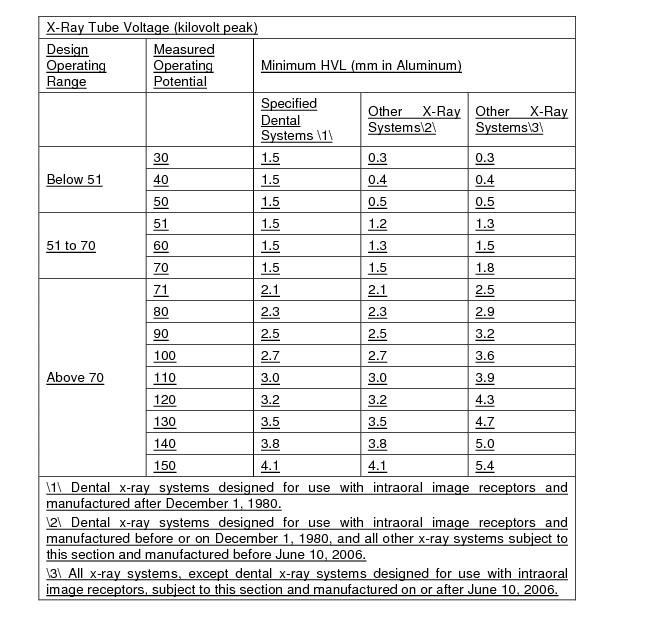
4.6.2 Optional filtration on fluoroscopic systems. Fluoroscopic systems manufactured on or after June 10, 2006, incorporating an x-ray tube(s) with a continuous output of 1 kilowatt or more and an anode heat storage capacity of 1 million heat units or more shall provide the option of adding x-ray filtration to the diagnostic source assembly in addition to the amount needed to meet the half-value layer provisions of this subsection. The selection of this additional x-ray filtration shall be either at the option of the user or automatic as part of the selected mode of operation. A means of indicating which combination of additional filtration is in the x-ray beam shall be provided. (21 CFR 1020.30(m)(2))
4.6.3 Measuring compliance. For capacitor energy storage equipment, compliance shall be determined with the maximum selectable quantity of charge per exposure.
4.7 Aluminum equivalent of material between patient and image receptor. Except when used in a CT x-ray system, the aluminum equivalent of each of the items listed in Table 2 in this paragraph, which are used between the patient and the image receptor, may not exceed the indicated limits. Compliance shall be determined by x-ray measurements made at a potential of 100 kilovolts peak and with an x-ray beam that has an HVL specified in Table 1 of this section for the potential. This requirement applies to front panel(s) of image receptors and film changers provided by the manufacturer for patient support or for prevention of foreign object intrusions. It does not apply to screens and their associated mechanical support panels or grids.
TABLE 2

4.8 Battery charge indicator. On battery-powered generators, visual means shall be provided on the control panel to indicate whether the battery is in a state of charge adequate for proper operation.
4.9 Modification of certified diagnostic x-ray components and systems.
4.9.1 Diagnostic x-ray components and systems certified in accordance with 21 CFR Part 1020 shall not be modified such that the component or system fails to comply with any applicable provision of this Part.
4.9.2 The owner of a diagnostic x-ray system who uses the system in a professional or commercial capacity may modify the system provided the modification does not result in the failure of the system or component to comply with the applicable requirements of this Part. The owner who causes such modification need not submit the reports required by this Part, provided the owner records the date and the details of the modification in the system records and maintains this information, and provided the modification of the x-ray system does not result in a failure to comply with this Part.
4.10 Multiple Tubes. Where two or more radiographic tubes are controlled by one exposure switch, the tube which has been selected shall be clearly indicated prior to initiation of the exposure. Only the selected tube can be energized. This indication shall be both on the x-ray control panel and at or near the tube housing assembly which has been selected.
4.11 Mechanical Support of Tube Head. The tube housing assembly supports shall be adjusted such that the tube housing assembly will remain stable during an exposure unless tube housing movement is a designed function of the x-ray system.
4.12 Locks. All position locking, holding, and centering devices on x-ray system components and systems shall function as intended.
4.13 Maintaining Compliance. Diagnostic x-ray systems and their associated components used on humans and certified pursuant to the Federal X-Ray Equipment Performance Standard (21 CFR Part 1020) shall be maintained in compliance with applicable requirements of that standard.
5.1 The provisions of this Part apply to equipment for fluoroscopic imaging or for recording images from the fluoroscopic image receptor. (21 CFR 1020.32)
5.2 Only image-intensified or direct-digital receptor fluoroscopic equipment shall be used for fluoroscopy.
5.3 Primary Protective Barrier.
5.3.1 Limitation of useful beam. The fluoroscopic imaging assembly shall be provided with a primary protective barrier which intercepts the entire cross section of the useful beam at any SID. The x-ray tube used for fluoroscopy shall not produce x-rays unless the barrier is in position to intercept the entire useful beam. The AKR due to transmission through the barrier with the attenuation block in the useful beam combined with radiation from the fluoroscopic imaging receptor shall not exceed 3.34x10-3 percent of the entrance AKR, at a distance of 10 cm from any accessible surface of the fluoroscopic imaging assembly beyond the plane of the image receptor. Radiation therapy simulation systems shall be exempt from this requirement provided the systems are intended only for remote control operation. (21 CFR 1020.32(a)(1))
5.3.2 Measuring compliance. The AKR shall be measured in accordance with Part F, subsection 5.6. The AKR due to transmission through the primary barrier combined with radiation from the fluoroscopic image receptor shall be determined by measurements averaged over an area of 100 square cm with no linear dimension greater than 20 cm. If the source is below the tabletop, the measurement shall be made with the input surface of the fluoroscopic imaging assembly positioned 30 cm above the tabletop. If the source is above the tabletop and the SID is variable, the measurement shall be made with the end of the beam-limiting device or spacer as close to the tabletop as it can be placed, provided that it shall not be closer than 30 cm. Movable grids and compression devices shall be removed from the useful beam during the measurement. For all measurements, the attenuation block shall be positioned in the useful beam 10 cm from the point of measurement of entrance AKR and between this point and the input surface of the fluoroscopic imaging assembly. (21CFR 1020.32(a)(2)).
5.4 Field Limitation.
5.4.1 Angulation. For fluoroscopic equipment manufactured after February 25, 1978, when the angle between the image receptor and the beam axis of the x-ray beam is variable, means shall be provided to indicate when the axis of the x-ray beam is perpendicular to the plane of the image receptor. Compliance with Part F, subsection 5.4.5 and Part F, subsection 5.4.6 shall be determined with the beam axis indicated to be perpendicular to the plane of the image receptor. (21 CFR 1020.32(b)(1))
5.4.2 Further means for limitation. Means shall be provided to permit further limitation of the x-ray field to sizes smaller than the limits of Part F, subsection 5.4.5 and Part F, subsection 5.4.6 Beam-limiting devices manufactured after May 22, 1979, and incorporated in equipment with a variable SID and/or capability of a visible area of greater than 300 cm2, shall be provided with means for stepless adjustment of the x-ray field. Equipment with a fixed SID and the capability of a visible area of no greater than 300 cm2 shall be provided with either stepless adjustment of the x-ray field or with a means to further limit the x-ray field size at the plane of the image receptor to 125 cm2 or less. Stepless adjustment shall, at the greatest SID, provide continuous field sizes from the maximum obtainable to a field size containable in a square of 5 cm by 5 cm. (21 CFR 1020.32(b)(2))
5.4.3 Spot-film devices. In addition to applicable regulations in Part F, Section 6.0 (Radiographic Equipment), the following requirements shall apply to spot-film devices, except when the spot-film device is provided for use with a radiation therapy simulation system: (21 CFR 1020.31(h))
5.4.3.1 Means shall be provided between the source and the patient for adjustment of the x-ray field size in the plane of the image receptor to the size of that portion of the image receptor which has been selected on the spot-film selector. Such adjustment shall be accomplished automatically when the x-ray field size in the plane of the image receptor is greater than the selected portion of the image receptor. If the x-ray field size is less than the size of the selected portion of the image receptor, the field size shall not open automatically to the size of the selected portion of the image receptor unless the operator has selected that mode of operation. (21 CFR 1020.31(h)(1))
5.4.3.2 Neither the length nor width of the x-ray field in the plane of the image receptor shall differ from the corresponding dimensions of the selected portion of the image receptor by more than 3 percent of the SID when adjusted for full coverage of the selected portion of the image receptor. The sum, without regard to sign, of the length and width differences shall not exceed 4 percent of the SID. On spot-film devices manufactured after February 25, 1978, if the angle between the plane of the image receptor and beam axis is variable, means shall be provided to indicate when the axis of the x-ray beam is perpendicular to the plane of the image receptor, and compliance shall be determined with the beam axis indicated to be perpendicular to the plane of the image receptor. (21 CFR 1020.31(h)(2))
5.4.3.3 The center of the x-ray field in the plane of the image receptor shall be aligned with the center of the selected portion of the image receptor to within 2 percent of the SID. (21 CFR 1020.31(h)(3))
5.4.3.4 Means shall be provided to reduce the x-ray field size in the plane of the image receptor to a size smaller than the selected portion of the image receptor such that: (21 CFR 1020.31(h)(4))
5.4.3.4.1 For spot-film devices used on fixed-SID fluoroscopic systems which are not required to, and do not provide stepless adjustment of the x-ray field, the minimum field size, at the greatest SID, does not exceed 125 square cm; or (21 CFR 1020.31(h)(4)(i))
5.4.3.4.2 For spot-film devices used on fluoroscopic systems that have a variable SID and/or stepless adjustment of the field size, the minimum field size, at the greatest SID, shall be containable in a square of 5 cm by 5 cm. (21 CFR 1020.31(h)(4)(ii))
5.4.4 A capability may be provided for overriding the automatic x-ray field size adjustment in case of system failure. If it is so provided, a signal visible at the fluoroscopist’s position shall indicate whenever the automatic x-ray field size adjustment override is engaged. Each such system failure override switch shall be clearly labeled as follows:
For X-ray Field Limitation System Failure
(21 CFR 1020.31(h)(5))
5.4.5 Fluoroscopy and radiography using the fluoroscopic imaging assembly with inherently circular image receptors.
5.4.5.1 For fluoroscopic equipment manufactured before June 10, 2006, other than radiation therapy simulation systems, the following applies: (21 CFR 1020.32(b)(4)(i)
5.4.5.1.1 Neither the length nor width of the x-ray field in the plane of the image receptor shall exceed that of the visible area of the image receptor by more than 3 percent of the SID. The sum of the excess length and the excess width shall be no greater than 4 percent of the SID. (21 CFR 1020.32(b)(4)(i)(A))
5.4.5.1.2 For rectangular x-ray fields used with circular image receptors, the error in alignment shall be determined along the length and width dimensions of the x-ray field which pass through the center of the visible area of the image receptor. (21 CFR 1020.32(b)(4)(i)(B))
5.4.5.2 For fluoroscopic equipment manufactured on or after June 10, 2006, other than radiation simulation systems, the maximum area of the x-ray field in the plane of the image receptor shall conform with one of the following requirements: (21 CFR 1020.32(b)(4)(ii))
5.4.5.2.1 When any linear dimension of the visible area of the image receptor measured through the center of the visible area is less than or equal to 34 cm in any direction, at least 80 percent of the area of the x-ray field overlaps the visible area of the image receptor, or (21 CFR 1020.32(b)(4)(ii)(A))
5.4.5.2.2 When any linear dimension of the visible area of the image receptor measured through the center of the visible area is greater than 34 cm in any direction, the x-ray field measured along the direction of greatest misalignment with the visible area of the image receptor does not extend beyond the edge of the visible area of the image receptor by more than 2 cm. (21 CFR 1020.32(b)(4)(ii)(B))
5.4.6 Fluoroscopy and radiography using fluoroscopic imaging assembly with inherently rectangular image receptors. For x-ray systems manufactured on or after June 10, 2006, the following applies: (21 CFR 1020.32(b)(5))
5.4.6.1 Neither the length nor width of the x-ray field in the plane of the image receptor shall exceed that of the visible area of the image receptor by more than 3 percent of the SID. The sum of the excess length and the excess width shall be no greater than 4 percent of the SID. (21 CFR 1020.32(b)(5)(i))
5.4.6.2 The error in alignment shall be determined along the length and width dimensions of the x-ray field which pass through the center of the visible area of the image receptor. (21 CFR 1020.32(b)(5)(ii))
5.4.7 Override capability. If the fluoroscopic x-ray field size is adjusted automatically as the SID or image receptor size is changed, a capability may be provided for overriding the automatic adjustment in case of system failure. If it is so provided, a signal visible at the fluoroscopist’s position shall indicate whenever the automatic field adjustment is overridden. Each such system failure override switch shall be clearly labeled as follows:
FOR X-RAY FIELD
LIMITATION SYSTEM FAILURE
(21 CFR 1020.32(b)(6))
5.5 Activation of Tube. X-ray production in the fluoroscopic mode shall be controlled by a device which requires continuous pressure by the operator for the entire time of any exposure. When recording serial radiographic images from the fluoroscopic image receptor, the operator shall be able to terminate the x-ray exposure(s) at any time, but means may be provided to permit completion of any single exposure of the series in process. (21 CFR 1020.32(c))
5.6 Air Kerma Rates. For fluoroscopic equipment, the following requirements apply:
5.6.1 Fluoroscopic equipment manufactured before May 19, 1995.
5.6.1.1 Equipment provided with automatic exposure rate control (AERC) shall not be operable at any combination of tube potential and current that will result in an AKR in excess of 88 mGy per minute (vice 10 R/min exposure rate) at the measurement point specified in Part F, subsection 5.6.4 except as specified in Part F, subsection 5.6.1.5. (21 CFR 1020.32(d)(1)(i))
5.6.1.2 Equipment provided without AERC shall not be operable at any combination of tube potential and current that will result in an AKR in excess of 44 mGy per minute (vice 5 R/min exposure rate) at the measurement point specified in Part F, subsection 5.6.5, except as specified in Part F, subsection 5.6.1.5. (21 CFR 1020.32(d)(1)(ii))
5.6.1.3 Equipment provided with both an AERC mode and a manual mode shall not be operable at any combination of tube potential and current that will result in an AKR in excess of 88 mGy per minute (vice 10 R/min exposure rate) in either mode at the measurement point specified in Part F, subsection 5.6.4, except as specified in Part F, subsection 5.6.1.5 (21 CFR 1020.32(d)(1)(iii))
5.6.1.4 Equipment may be modified in accordance with this Part to comply with Part F, subsection 5.6.2. When the equipment is modified, it shall bear a label indicating the date of the modification and the statement:
MODIFIED TO COMPLY WITH 21 CFR 1020.32(H)(2)
(21 CFR 1020.32(d)(1)(iv))
5.6.1.5 Exceptions: During recording of fluoroscopic images.
5.6.2 Fluoroscopic equipment manufactured on or after May 19, 1995.
5.6.2.1 Shall be equipped with AERC if operable at any combination of tube potential and current that results in an AKR greater than 44 mGy per minute (vice 5 R/min exposure rate) at the measurement point specified in Part F, subsection 5.6.4. Provision for manual selection of technique factors may be provided. (21 CFR 1020.32(d)(2)(i))
5.6.2.2 Shall not be operable at any combination of tube potential and current that will result in an AKR in excess of 88 mGy per minute (vice 10 R/min exposure rate) at the measurement point specified in Part F, subsection 5.6.4, except as specified in Part F subsection 5.6.2.3. (21 CFR 1020.32(d)(2)(ii))
5.6.2.3 Exceptions:
5.6.2.3.1 For equipment manufactured prior to June 10, 2006, during the recording of images from a fluoroscopic image receptor using photographic film or a video camera when the x-ray source is operated in a pulsed mode. (21 CFR 1020.32(d)(2)(iii)(A))
5.6.2.3.2 For equipment manufactured on or after June 10, 2006, during the recording of images from the fluoroscopic image receptor for the purpose of providing the user with a recorded image(s) after termination of the exposure. Such recording does not include images resulting from a last-image-hold feature that are not recorded. (21 CFR 1020.32(d)(2)(iii)(B))
5.6.3 Fluoroscopy equipment with optional high-level control
5.6.3.1 When high-level control is selected and the control is activated, in which case the equipment shall not be operable at any combination of tube potential and current that will result in an AKR in excess of 176 mGy per minute (vice 20 R/min exposure rate) at the measurement point specified in Part F, subsection 5.6.4. Special means of activation of high-level controls shall be required. The high-level control shall be operable only when continuous manual activation is provided by the operator. A continuous signal audible to the fluoroscopist shall indicate that the high-level control is employed.
5.6.4 Measuring compliance. Compliance with this subsection shall be determined as follows:
5.6.4.1 If the source is below the x-ray table, the AKR shall be measured at 1 cm above the tabletop or cradle. (21 CFR 1020.32(d)(3)(i))
5.6.4.2 If the source is above the x-ray table, the AKR shall be measured at 30 cm above the tabletop with the end of the beam-limiting device or spacer positioned as closely as possible to the point of measurement. (21 CFR 1020.32(d)(3)(ii))
5.6.4.3 In a C-arm type of fluoroscope, the AKR shall be measured at 30 cm from the input surface of the fluoroscopic imaging assembly, with the source positioned at any available SID, provided that the end of the beam-limiting device or spacer is no closer than 30 cm from the input surface of the fluoroscopic imaging assembly. (21 CFR 1020.32(d)(3)(iii))
5.6.4.4 In a C-arm type of fluoroscope having an SID less than 45 cm, the AKR shall be measured at the minimum SSD. (21 CFR 1020.32(d)(3)(iv))
5.6.4.5 In a lateral type of fluoroscope, the air kerma rate shall be measured at a point 15 cm from the centerline of the x-ray table and in the direction of the x-ray source with the end of the beam-limiting device or spacer positioned as closely as possible to the point of measurement. If the tabletop is movable, it shall be positioned as closely as possible to the lateral x-ray source, with the end of the beam-limiting device or spacer no closer than 15 cm to the centerline of the x-ray table. (21 CFR 1020.32(d)(3)(v))
5.6.5 Exemptions. Fluoroscopic radiation therapy simulation systems are exempt from the requirements set forth in Part F, subsection 5.6 when used for therapy simulation purposes. (21 CFR 1020.32(d)(4))
5.7 Indication of potential and current. During fluoroscopy and cinefluorography, x-ray tube potential and current shall be continuously indicated. Deviation of x-ray tube potential and current from the indicated value shall not exceed the maximum deviation as stated by the manufacturer. (21 CFR 1020.32(f))
5.8 Source-skin distance.
5.8.1 Means shall be provided to limit the source-skin distance to not less than 38 cm on stationary fluoroscopes and to not less than 30 cm on mobile and portable fluoroscopes. In addition, for fluoroscopes intended for specific surgical or interventional applications that would be prohibited at the source-skin distances specified in this paragraph, provisions may be made for operating at shorter source-skin distances but in no case less than 20 cm.
5.8.2 For stationary, mobile, or portable C-arm fluoroscopic systems manufactured on or after June 10, 2006, having a maximum source-image receptor distance of less than 45 cm, means shall be provided to limit the source-skin distance to not less than 19 cm. Such systems shall be labeled for extremity use only. In addition, for those systems intended for specific surgical that would be prohibited at the source-skin distance specified in this paragraph, provisions may be made for operation at shorter source-skin distances but in no case less than 10 cm.
5.9 Fluoroscopic irradiation time, display, and signal.
5.9.1 Fluoroscopic equipment manufactured before June 10, 2006:
5.9.1.1 Shall be provided with means to preset the cumulative irradiation time of the fluoroscopic tube. The maximum cumulative time of the timing device shall not exceed 5 minutes without resetting. A signal audible to the fluoroscopist shall indicate the completion of any preset cumulative irradiation time. Such signal shall continue to sound while x-rays are produced until the timing device is reset. Fluoroscopic equipment may be modified in accordance with 21 CFR 1020.30(q) to comply with the requirements of this paragraph. When the equipment is modified, it shall bear a label indicating the statement:
Modified to comply with 21 CFR 1020.32(h)(2)
(21 CFR 1020.32(h)(1)(i))
5.9.1.2 As an alternative to the requirements of this paragraph, radiation therapy simulation systems may be provided with a means to indicate the total cumulative exposure time during which x-rays were produced, and which is capable of being reset between x-ray examinations. (21 CFR 1020.32(h)(1)(ii))
5.9.2 For x-ray controls manufactured on or after June 10, 2006, there shall be provided for each fluoroscopic tube:
5.9.2.1 A display of the fluoroscopic irradiation time at the fluoroscopist’s working position. This display shall function independently of the audible signal described in this subsection. The following requirements apply: (variation of 21 CFR 1020.32(h)(2)(i))
5.9.2.1.1 When the x-ray tube is activated, the fluoroscopic irradiation time in minutes and tenths of minutes shall be continuously displayed and updated at least once every 6 seconds. (21 CFR 1020.32(h)(2)(i)(A))
5.9.2.1.2 The fluoroscopic irradiation time shall also be displayed within 6 seconds of termination of an exposure and remain displayed until reset. (21 CFR 1020.32(h)(2)(i)(B))
5.9.2.1.3 Means shall be provided to reset the display to zero prior to the beginning of a new examination or procedure. (21 CFR 1020.32(h)(2)(i)(C))
5.9.2.2 A signal audible to the fluoroscopist shall sound for each passage of 5 minutes of fluoroscopic irradiation time during an examination or procedure. The signal shall sound until manually reset or, if automatically reset, for at least 2 seconds. (21 CFR 1020.32(h)(2)(ii))
5.10 Display of last-image-hold (LIH). Fluoroscopic equipment manufactured on or after June 10, 2006, shall be equipped with means to display LIH image following termination of the fluoroscopic exposure. (21 CFR 1020.32(j))
5.10.1 For an LIH image obtained by retaining pretermination fluoroscopic images, if the number of images and method of combining images are selectable by the user, the selection shall be indicated prior to initiation of the fluoroscopic exposure. (21 CFR 1020.32(j)(1))
5.10.2 For an LIH image obtained by initiating a separate radiographic-like exposure at the termination of fluoroscopic imaging, the technique factors for the LIH image shall be selectable prior to the fluoroscopic exposure, and the combination selected shall be indicated prior to initiation of the fluoroscopic exposure. (21 CFR 1020.32(j)(2))
5.10.3 Means shall be provided to clearly indicate to the user whether a displayed image is the LIH radiograph or fluoroscopy. Display of the LIH radiograph shall be replaced by the fluoroscopic image concurrently with re-initiation of fluoroscopic exposure, unless separate displays are provided for the LIH radiograph and fluoroscopic images. (21 CFR 1020.32(j)(3))
5.11 Displays of values of AKR and cumulative air kerma. Fluoroscopic equipment manufactured on or after June 10, 2006, shall display at the fluoroscopist’s working position the AKR and cumulative air kerma. The following requirements apply for each x-ray tube used during an examination or procedure: (21 CFR 1020.32(k))
5.11.1 When the x-ray tube is activated and the number of images produced per unit time is greater than six images per second, the AKR in mGy/min shall be continuously displayed and updated at least once every second. (21 CFR 1020.32(k)(1))
5.11.2 The cumulative air kerma in units of mGy shall be displayed either within 5 seconds of termination of an exposure or displayed continuously and updated at least once every 5 seconds. (21 CFR 1020.32(k)(2))
5.11.3 The display of the AKR shall be clearly distinguishable from the display of the cumulative air kerma. (21 C FR 1020.32(k)(3))
5.11.4 The AKR and cumulative air kerma shall represent the value for conditions of free-in-air irradiation at one of the following reference locations specified according to the type of fluoroscope. (21 CFR 1020.32(k)(4))
5.11.4.1 For fluoroscopes with x-ray source below the x-ray table, x-ray source above the table, or of lateral type, the reference location shall be the respective locations specified in Part F, subsection 5.6.4.1, 5.6.4.2 or 5.6.4.5 (21 CFR 1020.32(k)(4)(i))
5.11.4.2 For C-arm fluoroscopes, the reference location shall be 15 cm from the isocenter toward the x-ray source along the beam axis. Alternatively, the reference location shall be at a point specified by the manufacturer to represent the location of the intersection of the x-ray beam with the patient’s skin. (21 CFR 1020.32(k)(4)(ii))
5.11.5 Means shall be provided to reset to zero the display of cumulative air kerma prior to the commencement of a new examination or procedure. (21 CFR 1020.32(k)(5))
5.11.6 The displayed AKR and cumulative air kerma shall not deviate from the actual values by more than ±35 percent over the range of 6 mGy/min and 100 mGy to the maximum indication of AKR and cumulative air kerma, respectively. Compliance shall be determined with an irradiation time greater than 3 seconds. (21 CFR 1020.32(k)(6))
5.12 Protection From Scatter Radiation.
5.12.1 For stationary fluoroscopic systems, ancillary shielding, such as drapes, self-supporting curtains, or viewing shields, shall be available and used as supplemental protection for all individuals other than the patient in the room during a fluoroscopy procedure.
5.12.1.2 Where sterile fields or special procedures prohibit the use of normal protective barriers or drapes, all of the following conditions shall be met.
5.12.1.2.1 Shielding required under Part F, subsection 5.12.1 shall be maintained to the degree possible under the clinical conditions.
5.12.1.2.2 All persons, except the patient, in the room where fluoroscopy is performed shall wear protective aprons that provide a lead equivalent shielding of at least 0.25 mm.
5.12.1.2.3 The fluoroscopic field size shall be reduced to the minimum required for the procedure being performed (area of clinical interest).
5.12.1.2.4 Operating and safety procedures shall reflect the above conditions, and fluoroscopy personnel shall exhibit awareness of situations requiring the use and/or non-use of the protective drapes.
5.13 Operator Qualifications.
5.13.1 In addition to the applicable sections of these regulations, the operation of a fluoroscopic x-ray system for clinical purposes shall be limited to:
5.13.1.1 A state licensed practitioner, or certified radiologic technologist working within his or her scope of practice;
5.13.1.2 A Radiologist Assistant (RA) (if recognized by the state licensing agency) working within his or her scope of practice and under the direct supervision of a licensed practitioner meeting the conditions of Part F, subsection 5.13.1.1;
5.13.1.3 A state-licensed physician’s assistant who passed the state-level American Registry of Radiologic Technologists (ARRT) Fluoroscopy Exam (or equivalent) and only under the direct supervision of the licensed practitioner meeting the conditions of Part F, subsection 5.13.1.1. This operator qualification is limited to physician’s assistants, and excludes nurse practitioners.
5.13.1.4 A medical resident or radiologic technology student, in training, and only under the personal supervision of the licensed practitioner meeting the conditions of Part F, subsection 5.13.1.1.
5.13.2 All persons operating, or supervising the operation of, fluoroscopy systems shall have completed training that includes but is not limited to the following:
5.13.2.1 Basic properties of radiation;
5.13.2.2 Biological effects of x-ray;
5.13.2.3 Radiation protection methods for patients and staff;
5.13.2.4 Units of measurement and dose, including DAP (dose-area product) values & air kerma;
5.13.2.5 Factors affecting fluoroscopic outputs;
5.13.2.6 High level control options;
5.13.2.7 Dose management including dose reduction techniques, monitoring, and recording;
5.13.2.8 Principles and operation of the specific fluoroscopic x-ray system(s) to be used;
5.13.2.9 Fluoroscopic and fluorographic outputs of each mode of operation on the system(s) to be used clinically; and
5.13.2.10 Applicable requirements of these regulations.
5.13.3 All persons operating, or supervising the operation of, fluoroscopy systems during Fluoroscopically Guided Interventional (FGI) procedures shall have completed training that includes but is not limited to:
5.13.3.1 The topics provided in Part F, subsection 5.13.2;
5.13.3.2 Methods to reduce patient dose using advanced imaging and recording features;
5.13.3.3 Procedures for recording pertinent data specified in Part F, subsection 5.16, and
5.13.3.4 Documentation pertaining to the requirements of Part F, Section 5.0 shall be maintained for review for three years.
5.14 Equipment Operation.
5.14.1 All fluoroscopic images shall be viewed, directly or indirectly, and interpreted by a licensed practitioner working within their scope of practice.
5.14.2 Overhead fluoroscopy shall not be used as a positioning tool for general purpose radiographic examinations.
5.14.3 Operators shall be competent in the standard operating procedures of the unit in use, including the use of available dose-saving features, and the relative radiation output rates of the various modes of operation.
5.14.4 Procedure planning for fluoroscopic procedures on pregnant patients shall include feasible modifications to minimize the dose to the conceptus.
5.14.5 Procedure planning for fluoroscopic procedures on pediatric patients shall include feasible modifications to minimize dose.
5.14.6 The registrant shall use all methods available on the fluoroscopy system to monitor dose during a fluoroscopic procedure.
5.14.7 The facility shall establish a written policy regarding patient dose management in fluoroscopically guided procedures in conformance with the ACR-AAPM Technical Standard for Management of the Use of Radiation in Fluoroscopic Procedures (ACR Resolution 44 – 2013), NCRP Report 168, or equivalent.
5.15 Qualified Medical Physicist Evaluations.
5.15.1 Fluoroscopic equipment shall be evaluated by a QMP within 30 days of installation. Any maintenance of the system that may affect the exposure rate shall be evaluated by a QMP, or internally authorized staff under the general direction of a QMP. Thereafter, the measurements shall be made of fluoroscopic equipment annually. At a minimum these evaluations shall include:
5.15.1.1 A measurement of entrance exposure rates that covers the full range of patient thicknesses, including those that are expected to drive the system to maximum output in all modes clinically used, including fluoroscopy, high-level control, acquisition, digital subtraction and Cineradiography, when available. These measurements shall:
5.15.1.1.1 For systems without automatic exposure control, be made utilizing a milliamperage and kVp typical of the clinical use of the fluoroscopic system;
5.15.1.1.2 For systems with automatic exposure control, be made utilizing sufficient attenuating material in the useful beam to produce a milliamperage and kVp typical of the clinical use of the fluoroscopic system;
5.15.1.2 A measurement and verification of compliance of maximum AKR for fluoroscopy and high-level control, if available. Measurements shall be made in accordance with Part F, subsection 5.6.4.
5.15.1.3 An evaluation of high contrast resolution and low contrast resolution in both fluoroscopic and spot-film modes.
5.15.1.4 An evaluation of the operation of the 5-minute timer, warning lights, interlocks, and collision sensors.
5.15.1.5 An evaluation of the beam quality and collimation in the fluoroscopy and spot-film modes.
5.15.1.6 An evaluation of the availability and accuracy of technique indicators and integrated radiation dose displays.
5.15.1.7 An evaluation of any changes that may impact patient and personnel protection devices.
5.15.2 Measurements required in Part F, subsection 5.15.1 shall be performed with a calibrated dosimetry system per manufacturer recommendations not to exceed 2 years and records maintained for 5 years for inspection by the Agency.
5.16 Additional requirements for facilities performing fluoroscopically-guided interventional (FGI) procedures.
5.16.1 A registrant utilizing FGI procedures shall establish a Radiation Protocol Committee (RPC) in accordance with the following.
5.16.1.1 The registrant may establish a system-wide committee if the registrant has more than one site.
5.16.1.2 Two or more registrants may form a cooperative RPC as long as each facility has a representative on the committee.
5.16.1.3 If the registrant has already established a radiation safety committee, the requirements of this subsection may be delegated to that committee if the members meet the requirements of Part F, subsection 5.16.5.
5.16.2 A quorum of the RPC shall meet as often as necessary, but at intervals not to exceed 12 months.
5.16.3 Record of RPC. A record of each RPC meeting shall include the date, names of individuals in attendance, minutes of the meeting, and any actions taken. The registrant shall maintain RPC meeting record for inspection by the Agency for at least three years.
5.16.4 Provide an annual report to the radiation safety committee, or to the radiation safety officer.
5.16.5 RPC Members. Members shall include but not be limited to the following individuals:
5.16.5.1 A supervising licensed practitioner of the healing arts who meets the requirements in Part F, subsection 5.13;
5.16.5.2 A QMP or QE;
5.16.5.3 A radiologic technologist; and
5.16.5.4 Other individuals as deemed necessary by the registrant. (eg. RSO, Chief Medical or Administrative Officer, Radiology Department Administrator/Manager)
5.16.6 Establish and implement FGI procedure protocols.
5.16.6.1 The RPC shall establish and implement written protocols, or protocols documented in an electronic report system, that include but are not limited to the following:
5.16.6.1.1 A method to be used to monitor patient radiation dose during FGI.
5.16.6.1.2 Dose notification levels, as appropriate, at which the physician is notified and appropriate actions are taken for patient safety.
5.16.6.1.3 Substantial Radiation Dose Level (SRDL) values following nationally recognized standards,
5.16.6.1.4 Actions to be taken for cases when a SRDL is exceeded which may include patient follow-up.
5.16.6.1.5 A review of the established protocols at an interval not to exceed 12 months.
5.16.6.2 A record of each RPC protocol shall be maintained for inspection by the Agency.
5.16.7 Procedures for maintaining records.
5.16.7.1 A record of radiation output information shall be maintained so the radiation dose to the skin may be estimated in accordance with established protocols. The record shall include the following:
5.16.7.1.1 Patient identification;
5.16.7.1.2 Type and date of examination;
5.16.7.1.3 Identification of the fluoroscopic system used; and
5.16.7.1.4 Peak skin dose, cumulative air kerma or dose area product used if the information is available on the fluoroscopic system.
5.16.7.1.5 If the peak skin dose, cumulative air kerma or dose area product are not displayed on the fluoroscopic system, records shall include other information necessary to estimate the radiation dose to the skin in accordance with established protocol or the following as necessary:
5.16.7.1.5.1 Fluoroscopic mode, such as, high-level or pulsed mode of operation;
5.16.7.1.5.2 Cumulative fluoroscopic exposure time; and
5.16.7.1.5.3 Number of films or recorded exposures.
5.16.7.2 The registrant shall maintain records required by this subparagraph for inspection by the Agency.
6.1 The following regulations apply to all non-dental registrants using diagnostic x-ray equipment. Requirements specific to using dental intra-oral, hand held, panoramic, and cephalometric equipment are in Part F, Section 7.0.
6.2 Digital radiographic systems shall be evaluated by a QMP (QE) prior to clinical use, and at least annually. The evaluation shall follow nationally recognized standards or procedures. Unless otherwise specified in this Part, dental, podiatric, and veterinary systems are exempt from this requirement.
6.3 Control and indication of technique factors.
6.3.1 Timers. Means shall be provided to terminate the exposure at a preset time interval, a preset product of current and time, a preset number of pulses, or a preset radiation exposure to the image receptor. (21 CFR 1020.31(a)(2))
6.3.1.1 Except during serial radiography, the operator shall be able to terminate the exposure at any time during an exposure of greater than one-half second. Except during panoramic dental radiography, termination of exposure shall cause automatic resetting of the timer to its initial setting or to zero. It shall not be possible to make an exposure when the timer is set to a zero or off position if either position is provided. (21 CFR 1020.31(a)(2)(i))
6.3.1.2 During serial radiography, the operator shall be able to terminate the x-ray exposure(s) at any time, but means may be provided to permit completion of any single exposure of the series in process. (21 CFR 1020.31(a)(2)(ii))
6.3.2 Automatic exposure controls. When an automatic exposure control is provided:
6.3.2.1 Indication shall be made on the control panel when this mode of operation is selected; (21 CFR 1020.31(a)(3)(i))
6.3.2.2 When the x-ray tube potential is equal to or greater than 51 kilovolts peak (kVp), the minimum exposure time for field emission equipment rated for pulse operation shall be equal to or less than a time interval equivalent to two pulses and the minimum exposure time for all other equipment shall be equal to or less than 1/60 second or a time interval required to deliver 5 milliampere-seconds (mAs), whichever is greater; (21 CFR 1020.31(a)(3)(ii))
6.3.2.3 Either the product of peak x-ray tube potential, current, and exposure time shall be limited to not more than 60 kilowatt-seconds (kWs) per exposure or the product of x-ray tube current and exposure time shall be limited to not more than 600 mAs per exposure, except when the x-ray tube potential is less than 51 kVp, in which case the product of x-ray tube current and exposure time shall be limited to not more than 2,000 mAs per exposure; and (21 CFR 1020.31(a)(3)(iii))
6.3.2.4 A visible signal shall indicate when an exposure has been terminated at the limits described in Part F, subsection 6.3.2.3, and manual resetting shall be required before further automatically timed exposures can be made. (21 CFR 1020.31(a)(3)(iv))
6.3.3 Accuracy. Deviation of technique factors under Part F, subsection 6.3 from indicated values shall not exceed the limits given by the manufacturer. (variation of 21 CFR 1020.31(a)(4))
6.4 Reproducibility.
6.4.1 Coefficient of variation. For any specific combination of selected technique factors, the estimated coefficient of variation of the air kerma shall be no greater than 0.05. (21 CFR 1020.31(b)(1)
6.4.2 Measuring compliance. Determination of compliance shall be based on 10 consecutive measurements taken within a time period of 1 hour. Equipment manufactured after September 5, 1978, shall be subject to the additional requirement that all variable controls for technique factors shall be adjusted to alternate settings and reset to the test setting after each measurement. The percent line-voltage regulation shall be within ±1 of the mean value for all measurements. For equipment having automatic exposure controls, compliance shall be determined with a sufficient thickness of attenuating material in the useful beam such that the technique factors can be adjusted to provide individual exposures of a minimum of 12 pulses on field emission equipment rated for pulsed operation or no less than one-tenth second per exposure on all other equipment. (21 CFR 1020.31(b)(2))
6.5 Linearity. The following requirements apply for any fixed x-ray tube potential within the range of 40 percent to 100 percent of the maximum rated. (variation of 21 CFR 1020.31(c))
6.5.1 Equipment having independent selection of x-ray tube current (mA). The average ratios of air kerma to the indicated milliampere-seconds product (mGy/mAs) obtained at any two consecutive tube current settings shall not differ by more than 0.10 times their sum. This is: ׀X1 – X2׀ ≤ 0.10(X1 + X2); where X1 and X2 are the average mGy/mAs values obtained at each of two consecutive mAs selector settings or at two settings differing by no more than a factor of 2 where the mAs selector provides continuous selection. (21 CFR 1020.31(c)(1))
6.5.2 Equipment having selection of x-ray tube current-exposure time product (mAs). For equipment manufactured after May 3, 1994, the average ratios of air kerma to the indicated milliampere-seconds product (mGy/mAs) obtained at any two consecutive mAs selector settings shall not differ by more than 0.10 times their sum. This is: ׀X1 – X2׀ ≤ 0.10(X1 + X2); where X1 and X2 are the average mGy/mAs values obtained at each of two consecutive mAs selector settings or at two settings differing by no more than a factor of 2 where the mAs selector provides continuous selection. (21 CFR 1020.31(c)(2))
6.5.3 Measuring compliance. Determination of compliance will be based on 10 exposures, made within 1 hour, at each of the two settings. These two settings may include any two focal spot sizes except where one is equal to or less than 0.45 mm and the other is greater than 0.45 mm. For purposes of this requirement, focal spot size is the focal spot size specified by the x-ray tube manufacturer. The percent line-voltage regulation shall be determined for each measurement. All values for percent line-voltage regulation at any one combination of technique factors shall be within ±1 of the mean value for all measurements at these technique factors. (21 CFR 1020.31(c)(3))
6.6 Field limitation and alignment for mobile, portable, and stationary general purpose x-ray systems. Except when spot-film devices are in service, mobile, portable, and stationary general purpose radiographic x-ray systems shall meet the following requirements: (21 CFR 1020.31(d))
6.6.1 Variable x-ray field limitation. A means for stepless adjustment of the size of the x-ray field shall be provided. Each dimension of the minimum field size at an SID of 100 cm shall be equal to or less than 5 cm. (21 CFR 1020.31(d)(1))
6.6.2 Visual definition.
6.6.2.1 Means for visually defining the perimeter of the x-ray field shall be provided. The total misalignment of the edges of the visually defined field with the respective edges of the x-ray field along either the length or width of the visually defined field shall not exceed 2 percent of the distance from the source to the center of the visually defined field when the surface upon which it appears is perpendicular to the axis of the x-ray beam. (21 CFR 1020.31(d)(2)(i))
6.6.2.2 When a light localizer is used to define the x-ray field, it shall provide an average illuminance of not less than 160 lux (15 footcandles) at 100 cm or at the maximum SID, whichever is less. The average illuminance shall be based on measurements made in the approximate center of each quadrant of the light field. Radiation therapy simulation systems are exempt from this requirement. (21 CFR 1020.31(d)(2)(ii))
6.6.2.3 The edge of the light field at 100 cm or at the maximum SID, whichever is less, shall have a contrast ratio, corrected for ambient lighting, of not less than 4 in the case of beam-limiting devices designed for use on stationary equipment, and a contrast ratio of not less than 3 in the case of beam-limiting devices designed for use on mobile and portable equipment. The contrast ratio is defined as I1/I2, where I1 is the illuminance 3 mm from the edge of the light field toward the center of the field; and I2 is the illuminance 3 mm from the edge of the light field away from the center of the field. Compliance shall be determined with a measuring aperture of 1 mm. (21 CFR 1020.31(d)(2)(iii))
6.7 Field indication and alignment on stationary general purpose x-ray equipment. Except when spot-film devices are in service, stationary general purpose x-ray systems shall meet the following requirements in addition to those prescribed in Part F, subsection 6.6: (21 CFR 1020.31(e))
6.7.1 Means shall be provided to indicate when the axis of the x-ray beam is perpendicular to the plane of the image receptor, to align the center of the x-ray field with respect to the center of the image receptor to within 2 percent of the SID, and to indicate the SID to within 2 percent; (21 CFR 1020.31(e)(1))
6.7.2 The beam-limiting device shall numerically indicate the field size in the plane of the image receptor to which it is adjusted; (21 CFR 1020.31(e)(2))
6.7.3 Indication of field size dimensions and SIDs shall be specified in centimeters and/or inches and shall be such that aperture adjustments result in x-ray field dimensions in the plane of the image receptor which correspond to those indicated by the beam-limiting device to within 2 percent of the SID when the beam axis is indicated to be perpendicular to the plane of the image receptor; and (21 CFR 1020.31(e)(3))
6.7.4 Compliance measurements will be made at discrete SIDs and image receptor dimensions in common clinical use (such as SIDs of 100, 150, and 200 cm and/or 36, 40, 48, 72 inches and nominal image receptor dimensions of 13, 18, 24, 30, 35, 40, and 43 cm and/or 5, 7, 8, 9, 10, 11, 12, 14, and 17 inches) or at any other specific dimensions at which the beam-limiting device or its associated diagnostic x-ray system is uniquely designed to operate. (21 CFR 1020.31(e)(4))
6.8 Field limitation on x-ray equipment other than general purpose radiographic systems.
6.8.1 X-ray systems designed for one image receptor size. Radiographic equipment designed for only one image receptor size at a fixed SID shall be provided with means to limit the field at the plane of the image receptor to dimensions no greater than those of the image receptor, and to align the center of the x-ray field with the center of image receptor to within 2 percent of the SID, or shall be provided with means to both size and align the x-ray field such that the x-ray field at the plane of the image receptor does not extend beyond the edge of the image receptor.
6.8.2 Other x-ray systems. Radiographic systems not specifically covered in Part F, subsection 6.7, subsection 6.8.2, subsection 6.8.3, and systems covered in Part F, subsection 6.8.1, which are also designed for use with extraoral image receptors and when used with an extraoral image receptor, shall be provided with means to limit the x-ray field in the plane of the image receptor so that such field does not exceed each dimension of the image receptor by more than 2 percent of the SID, when the axis of the x-ray beam is perpendicular to the plane of the image receptor. In addition, means shall be provided to align the center of the x-ray field with the center of the image receptor to within 2 percent of the SID, or means shall be provided to both size and alignment the x-ray field such that the x-ray field at the plane of the image receptor does not extend beyond any edge of the image receptor. These requirements may be met with: (21 CFR 1020.31(f)(4))
6.8.2.1 A system which performs in accordance with Part F, subsection 6.6 and Part F, subsection 6.7; or when alignment means are also provided, may be met with either; (21 CFR 1020.31(f)(4)(i))
6.8.2.2 An assortment of removable, fixed-aperture, beam-limiting devices sufficient to meet the requirement for each combination of image receptor size and SID for which the unit is designed. Each such device shall have clear and permanent markings to indicate the image receptor size and SID for which it is designed; or (21 CFR 1020.31(f)(4)(ii))
6.8.2.3 A beam-limiting device having multiple fixed apertures sufficient to meet the requirement for each combination of image receptor size and SID for which the unit is designed. Permanent, clearly legible markings shall indicate the image receptor size and SID for which each aperture is designed and shall indicate which aperture is in position for use. (21 CFR 1020.31(f)(4)(iii))
6.9 Positive beam limitation (PBL). The requirements of this subsection shall apply to radiographic systems which contain PBL. (21 CFR 1020.31(g))
6.9.1 Field size. When a PBL system is provided, it shall prevent x-ray production when: (21 CFR 1020.31(g)(1))
6.9.1.1 Either the length or width of the x-ray field in the plane of the image receptor differs from the corresponding image receptor dimension by more than 3 percent of the SID; or (21 CFR 1020.31(g)(1)(i))
6.9.1.2 The sum of the length and width differences stated in Part F, subsection 6.9.1.1 without regard to sign exceeds 4 percent of the SID. (21 CFR 1020.31(g)(1)(ii))
6.9.1.3 The beam-limiting device is at an SID for which PBL is not designed for sizing. (21 CFR 1020.31(g)(1)(iii))
6.9.2 Conditions for Positive Beam Limitation (PBL). When provided, the PBL system shall function as described in Part F, subsection 6.9.1 whenever all the following conditions are met: (21 CFR 1020.31(g)(2))
6.9.2.1 The image receptor is inserted into a permanently mounted cassette holder; (21 CFR 1020.31(g)(2)(i))
6.9.2.2 The image receptor length and width are less than 50 cm; (21 CFR 1020.31(g)(2)(ii))
6.9.2.3 The x-ray beam axis is within ±3 degrees of vertical and the SID is 90 cm to 130 cm inclusive; or the x-ray beam axis is within ±3 degrees of horizontal and the SID is 90 cm to 205 cm inclusive; (21 CFR 1020.31(g)(2)(iii))
6.9.2.4 The x-ray beam axis is perpendicular to the plane of the image receptor to within ±3 degrees; and (21 CFR 1020.31(g)(2)(iv))
6.9.2.5 Neither tomographic nor stereoscopic radiography is being performed. (21 CFR 1020.31(g)(2)(v))
6.9.3 Measuring compliance. Compliance with the requirements of Part F, subsection 6.9.1 shall be determined when the equipment indicates that the beam axis is perpendicular to the plane of the image receptor and the provisions of Part F, subsection 6.9.2 are met. Compliance shall be determined no sooner than 5 second after insertion of the image receptor. (21 CFR 1020.31(g)(3))
6.9.4 Operator initiated undersizing. The PBL system shall be capable of operating such that, at the discretion of the operator, the size of the field may be made smaller than the size of the image receptor through stepless adjustment of the field size. Each dimension of the minimum field size at an SID of 100 cm shall be equal to or less than 5 cm. Return to PBL function as described in Part F, subsection 6.9.1 shall occur automatically upon any change of image receptor size or SID. (21 CFR 1020.31(g)(4))
6.9.5 Override of PBL. A capability may be provided for overriding PBL in case of system failure and for servicing the system. This override may be for all SIDs and image receptor sizes. A key shall be required for any override capability that is accessible to the operator. It shall not be possible to remove the key while PBL is overridden. Each such key switch or key shall be clearly and durably labeled as follows:
For X-Ray Field Limitation System Failure
The override capability is considered accessible to the operator if it is referenced in the operator’s manual or in other material intended for the operator or if its location is such that the operator would consider it part of the operational controls. (21 CFR 1020.31(g)(5))
6.8.6 Disabling of PBL. A facility has the option to permanently functionally disable a PBL system. When this option is chosen, the standards for manual collimation apply.
6.10 Source-skin distance. The minimum source-skin distance shall not be less than 30 cm, except intraoral dental equipment covered under Part F, subsection 7.17.2 and veterinary equipment.
6.11 Radiation from capacitor energy storage equipment. Radiation emitted from the x-ray tube shall not exceed: (21 CFR 1020.31(l))
6.11.1 An air kerma of 0.26 microGy (vice 0.03 mR exposure) in 1 minute at 5 cm from any accessible surface of the diagnostic source assembly, with the beam-limiting device fully open, the system fully charged, and the exposure switch, timer, or any discharge mechanism not activated. Compliance shall be determined by measurements averaged over an area of 100 square cm, with no linear dimensions greater than 20 cm; and (21 CFR 1020.31(l)(1))
6.11.2 An air kerma of 0.88 mGy (vice 100 mR exposure) in one hour at 100 cm from the x-ray source, with beam-limiting device fully open, when the system is discharged through the x-ray tube either manually or automatically by use of a discharge switch or deactivation of the input power. Compliance shall be determined by measurements of the maximum air kerma per discharge multiplied by the total number of discharges in 1 hour (duty cycle). The measurements shall be averaged over an area of 100 square cm with no linear dimension greater than 20 cm. (21 CFR 1020.31(l)(2))
6.12 Radiation Exposure Control.
6.12.1 Exposure Initiation. Means shall be provided to initiate the radiation exposure by a deliberate action on the part of the operator, such as the depression of a switch. Radiation exposure shall not be initiated without such an action. In addition, it shall not be possible to initiate an exposure when the timer is set to a "zero" or "off" position if either position is provided.
6.12.2 Exposure Indication. Means shall be provided for visual indication observable at or from the operator's protected position whenever x-rays are produced. In addition, a signal audible to the operator shall indicate that the exposure has terminated.
6.12.3 Operator Protection, Except Veterinary Systems.
6.12.3.1 Stationary Radiographic Systems. Stationary radiographic systems shall be required to have the x-ray control, including the exposure switch, permanently mounted in a protected area so that the operator is required to remain in that protected area during the entire exposure.
6.12.3.2 Mobile and Portable Systems. Mobile and portable x-ray systems which are:
6.12.3.2.1 Used continuously for greater than one week in the same location, i.e., a room or suite, shall meet the requirements of Part F, subsection 6.12.3.1;
6.12.3.2.2 Used for less than one week at the same location shall be provided with either a protective barrier at least 2 meters (6.5 feet) high for operator protection during exposures, or means shall be provided to allow the operator to be at least 2.7 meters (9 feet) from the tube housing assembly during the exposure.
6.11.3.2.3 Podiatry Systems. Podiatry facilities shall meet the protection requirements in Part F, subsection 6.12.3.2.2.
6.12.4 Operator and Ancillary Personnel Protection for Veterinary Systems. All stationary, mobile or portable x-ray systems used for veterinary work shall be provided with either a 2 meter (6.5 feet) high protective barrier for operator protection during exposures, or shall be provided with means to allow the operator to be at least 2 meters (6.5 feet) from the tube housing assembly during exposures. Otherwise, in cases where animals are held, the operator and ancillary personnel shall be protected by a minimum of 0.25 mm lead equivalent from scatter radiation and 0.5 mm from the useful beam. Refer to Part F, Section 7.0 for hand-held intraoral dental x-ray units used in veterinary practice.
6.13 Tube Stands for Portable X-Ray Systems. Except during veterinary field operations where it is impractical to do so, a tube stand or other mechanical support shall be used for portable x-ray systems, so that the x-ray tube housing assembly need not be hand-held during an exposure.
6.14 Systems designed for mammography. All systems designed for mammography shall comply with Mammography Quality Standards Act of 1998.
6.15 Prohibitions. Capacity energy storage equipment shall not be used to image humans 2 years after the effective date of this Part.
7.1 In addition to the applicable provisions of Part F, Section 3.0, the requirements of Part F, Section 7.0 apply to dental facilities using intraoral, panoramic, and cephalometric x-ray equipment. Dental facilities using cone beam computed tomography (CBCT) technology shall follow applicable provisions of Part F, subsection 11.8.
7.2 Quality Assurance. In addition to the general quality assurance provisions in Part F, Section 3.0, the following requirements apply to a dental facility:
7.2.1 If using film, maintain a light-tight darkroom, use proper safelighting and safeguards, and evaluate darkroom integrity and daylight loading systems for film fog every six months and after a change that may impact film fog.
7.2.2 If using a filmless system, maintain and operate PSP and DDR systems according to manufacturer specifications.
7.2.3 Registrant shall provide initial orientation to x-ray operators to include but not limited to: positioning of the x-ray tube, image processing, operator location during x-ray exposure, source to skin distance, radiation protection, appropriate radiographic protocol, and applicable regulatory requirements. Records of training shall be maintained for inspection by the Agency.
7.3 Warning Label.
7.3.1 On systems manufactured on or before June 10, 2006, the control panel containing the main power switch shall bear the warning statement or the warning statement in Part F, subsection 7.3.2, legible and accessible to view: "WARNING: This x-ray unit may be dangerous to patient and operator unless safe exposure factors, operating instructions are observed."
7.3.2 On systems manufactured after June 10, 2006, the control panel containing the main power switch shall bear the warning statement, legible and accessible to view: "WARNING: This x-ray unit may be dangerous to patient and operator unless safe exposure factors, operating instructions and maintenance schedules are observed."
7.4 Radiation Exposure Control. Means shall be provided to initiate the radiation exposure by a deliberate action on the part of the operator, such as the depression of a switch. Radiation exposure shall not be initiated without such an action.
7.5 Exposure Control Location and Operator Protection. Except for units designed to be hand-held, the exposure control shall allow the operator to be:
7.5.1 Behind a protective barrier at least 2 meters (6.5 feet) tall or
7.5.2 At least 2 meters (6.5 feet) from the tube housing assembly, outside the path of the useful x-ray beam, while making exposures.
7.6 Administrative Controls.
7.6.1 Patient and image receptor holding devices shall be used when the techniques permit.
7.6.2 Except for units designed to be hand-held, the tube housing and position indicating device (PID) shall not be hand-held during an exposure.
7.6.3 Dental fluoroscopy without image intensification shall not be used.
7.7 Hand-Held Intraoral Equipment. In addition to the standards in this chapter, the following applies specifically to hand-held devices:
7.7.1 The hand-held x-ray system shall be equipped with a backscatter shield of not less than 0.25 mm lead equivalent and 15.2 cm (6 inches) in diameter that is positioned as close as practicable to the distal end of the position indication device.
7.7.2 The facility shall maintain documentation that each operator has completed training as specified by the manufacturer, and approved by the Agency.
7.7.3 The facility shall adopt and follow protocols provided by the manufacturer, and approved by the agency, regarding the safe operation of the device.
7.7.4 When operating a hand-held intraoral dental radiographic unit, operators shall wear a 0.25 mm lead equivalent apron, unless otherwise authorized by the Agency or a certified health or qualified medical physicist.
7.7.5 If the operator has difficulty in holding the device stationary during the exposure, the operator shall use a stand to immobilize the device.
7.7.6 The registrant shall secure the hand-held device from unauthorized removal or use, and report any lost or stolen device to the Agency within 24 hours of discovery.
7.7.7 The registrant shall maintain a Usage Log listing every procedure performed on a patient, to include user name, date, time of use. Usage Log shall be available for inspection by the Agency.
7.8 Beam-on indicators. The x-ray control shall provide visual indication whenever x-rays are produced. In addition, a signal audible to the operator shall indicate that the exposure has terminated. (21 CFR 1020.31(j))
7.9 Multiple Tubes. Where two or more radiographic tubes are controlled by one exposure switch, the tube which has been selected shall be clearly indicated prior to initiation of the exposure. Only the selected tube can be energized. This indication shall be both on the x-ray control panel and at or near the tube housing assembly which has been selected. (21 CFR 1020.31(k))
7.10 Mechanical Support of Tube Head. The tube housing assembly supports shall be adjusted such that the tube housing assembly will remain stable during an exposure unless tube housing movement is a designed function of the x-ray system.
7.11 Battery charge indicator. On battery-powered generators, visual means shall be provided on the control panel to indicate whether the battery is in a state of charge adequate for proper operation. (21 CFR 1020.30(o))
7.12 Locks. All position locking, holding, and centering devices on x-ray system components and systems shall function as intended.
7.13 Technique Indicators.
7.13.1 For x-ray equipment capable of displaying technique factors, the technique factors to be used during an exposure shall be indicated before the exposure begins. If automatic exposure controls are used, the technique factors which are set prior to the exposure shall be indicated. (21 CFR 1020.31(a)(1))
7.13.2 The requirement of Part F, subsection 7.13.1 may be met by permanent markings on equipment having fixed technique factors. (21 CFR 1020.31(a)(1))
7.14 Exposure Reproducibility. For any specific combination of selected technique factors, the estimated coefficient of variation of the air kerma shall be no greater than 0.05. (21 CFR 1020.31(b)(1))
7.15 Timers. Means shall be provided to terminate the exposure at a preset time interval, a preset product of current and time, a preset number of pulses, or a preset radiation exposure to the image receptor. (21 CFR 1020.31(a)(2))
7.16 Kilovolt Peak. Deviation of technique factors from indicated values shall not exceed the limits provided by the manufacturer. (variation of 21 CFR 1020.31(a)(4)) At a minimum, the kVp on variable kVp units shall be accurate to within 10 percent and within 20 percent on fixed kVp units.
7.17 X-ray Beam Alignment.
7.17.1 The useful x-ray beam shall be limited to the area of clinical interest.
7.17.2 Intraoral Dental Units
7.17.2.1 X-ray systems designed for use with an intraoral image receptor shall be provided with means to limit the source-to-skin distance (SSD) to not less than 18 cm (21 CFR1020.31(i)(1))
7.17.2.2 The x-ray field at the minimum SSD shall be containable in a circle having a diameter of no more than 7 cm. (21 CFR 1020.31(f)(1)(i))
7.17.3 Extraoral, Panoramic and Cephalometric Units
7.17.3.1 X-ray systems designed for use with extraoral image receptors and when used with an extraoral image receptor, shall be provided with means to limit the x-ray field in the plane of the image receptor so that such field does not exceed each dimension of the image receptor by more than 2 percent of the SID, when the axis of the x-ray beam is perpendicular to the plane of the image receptor. In addition, means shall be provided to align the center of the x-ray field with the center of the image receptor to within 2 percent of the SID, or means shall be provided to both size and alignment the x-ray field such that the x-ray field at the plane of the image receptor does not extend beyond any edge of the image receptor. These requirements may be met with: (21 CFR 1020.31(f)(4))
7.17.3.1.1 An assortment of removable, fixed-aperture, beam-limiting devices sufficient to meet the requirement for each combination of image receptor size and SID for which the unit is designed. Each such device shall have clear and permanent markings to indicate the image receptor size and SID for which it is designed; or (21 CFR 1020.31(f)(4)(ii))
7.17.3.1.2 A beam-limiting device having multiple fixed apertures sufficient to meet the requirement for each combination of image receptor size and SID for which the unit is designed. Permanent, clearly legible markings shall indicate the image receptor size and SID for which each aperture is designed and shall indicate which aperture is in position for use. (21 CFR 1020.31(f)(4)(iii))
7.18 Beam Quality. The Half Value Layer (HVL) of the useful beam for a given x-ray tube potential shall not be less than the values shown in Table 1. If it is necessary to determine such half-value layer at an x-ray tube potential which is not listed in Table 1 of this section, linear interpolation or extrapolation may be made. Positive means shall be provided to ensure that at least the minimum filtration needed to achieve beam quality requirements is in the useful beam during each exposure. In the case of a system, which is to be operated with more than one thickness of filtration, this requirement can be met by a filter interlocked with the kilovoltage selector which will prevent x-ray emissions if the minimum required filtration is not in place. (21 CFR 1020.30)
Table 1
(21 CFR 1020.30(m))
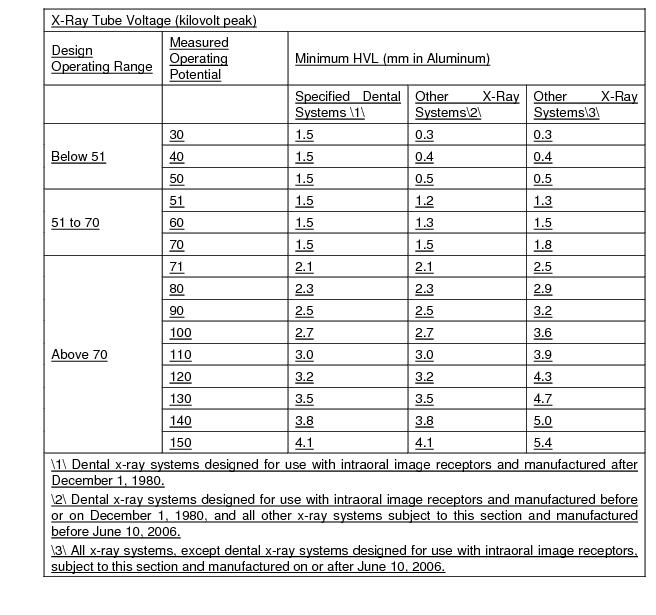
7.19 Intraoral dental x-ray machines shall not be operated at less than a measured 51 kVp effective 2 years after the publication of this rule.
7.20 Modification of certified diagnostic x-ray components and systems.
7.20.1 Diagnostic x-ray components and systems certified in accordance with 21 CFR Part 1020 shall not be modified such that the component or system fails to comply with any applicable provision of this Part. (21 CFR 1020.30(q) but doesn’t mention variance option)
7.20.2 The owner of a diagnostic x-ray system who uses the system in a professional or commercial capacity may modify the system provided the modification does not result in the failure of the system or component to comply with the applicable requirements of this Part. The owner who causes such modification need not submit the reports required by this Part, provided the owner records the date and the details of the modification in the system records and maintains this information, and provided the modification of the x-ray system does not result in a failure to comply with this Part. (21 CFR 1020.30(q)(2))
7.21 Leakage Radiation from the Diagnostic Source Assembly. The leakage radiation from the diagnostic source assembly measured at a distance of 1 meter in any direction from the source shall not exceed 0.88 milligray (mGy) air kerma (vice 100 milliroentgen (mR) exposure) in 1 hour when the x-ray tube is operated at its leakage technique factors. If the maximum rated peak tube potential of the tube housing assembly is greater than the maximum rated peak tube potential for the diagnostic source assembly, positive means shall be provided to limit the maximum x-ray tube potential to that of the diagnostic source assembly. Compliance shall be determined by measurements averaged over an area of 100 square centimeters with no linear dimension greater than 20 centimeters. (21 CFR 1020.30(k))
7.22 Radiation from Components Other Than the Diagnostic Source Assembly. The radiation emitted by a component other than the diagnostic source assembly shall not exceed an air kerma of 18 microgray (vice 2 milliroentgens exposure) in 1 hour at 5 centimeters from any accessible surface of the component when it is operated in an assembled x-ray system under any conditions for which it was designed. Compliance shall be determined by measurements averaged over an area of 100 square centimeters with no linear dimension greater than 20 centimeters. (21 CFR 1020.30(l))
7.23 Maintaining Compliance. Diagnostic x-ray systems and their associated components used on humans and certified pursuant to the Federal X-Ray Equipment Performance Standard (21 CFR Part 1020) shall be maintained in compliance with applicable requirements of that standard.
11.1 Requirements for CT Equipment.
11.1.1 Accreditation. All diagnostic CT x-ray systems for human use should be accredited by a nationally recognized accrediting organization.
11.1.2 Technical and Safety Information. The technical and safety information relating to the conditions of operation, dose information and imaging performance provided by the CT manufacturer shall be maintained by the facility.
11.1.3 Termination of Exposure.
11.1.3.1 Means shall be provided to terminate the x-ray exposure automatically by either de-energizing the x-ray source or shuttering the x-ray beam in the event of equipment failure affecting data collection. Such termination shall occur within an interval that limits the total scan time to no more than 110 percent of its preset value through the use of either a backup timer or devices which monitor equipment function. (21 CFR 1020.33(f)(2)(i))
11.1.3.2 A visible signal shall indicate when the x-ray exposure has been terminated through the means required by Part F, subsection 11.1.3.1. (21 CRF 1020.33(f)(2)(i))
11.1.3.3 The operator shall be able to terminate the x-ray exposure at any time during a scan, or series of scans under CT x-ray system control, of greater than one-half second duration. (first part of 21 CFR 1020.33(f)(2)(ii))
11.1.4 Tomographic Plane Indication and Alignment.
11.1.4.1 For any single tomogram system, means shall be provided to permit visual determination of the tomographic plane or a reference plane offset from the tomographic plane. (21 CFR 1020.33(g)(1))
11.1.4.2 For any multiple tomogram system, means shall be provided to permit visual determination of the location of a reference plane. This reference plane can be offset from the location of the tomographic planes. (version of 21 CFR 1020.33(g)(2))
11.1.4.3 If a mechanism using a light source is used to satisfy the requirements of Subsections Part F, subsection 11.1.4.1 or Part F, subsection 11.1.4.2, the light source shall allow visual determination of the location of the tomographic plane or reference plane under ambient light conditions of up to 500 lux. (21 CFR 1020.33(g)(5))
11.1.5 Beam-On and Shutter Status Indicators and Control Switches.
11.1.5.1 The CT x-ray control and gantry shall provide visual indication whenever x-rays are produced and, if applicable, whether the shutter is open or closed. (First part of 21 CFR 1020.33(h)(1))
11.1.5.2 Each emergency button or switch shall be clearly labeled as to its function.
11.1.6 Indication of CT Conditions of Operation.
11.1.6.1 The CT x-ray system shall be designed such that the CT conditions of operation to be used during a scan or a scan sequence shall be indicated prior to the initiation of a scan or a scan sequence. On equipment having all or some of these conditions of operation at fixed values, this requirement may be met by permanent markings. Indication of CT conditions of operation shall be visible from any position from which scan initiation is possible. (21 CFR 1020.33(f))
11.1.7 Additional Requirements Applicable to CT X-Ray Systems Containing a Gantry Manufactured After September 3, 1985.
11.1.7.1 The total error in the indicated location of the tomographic plane or reference plane shall not exceed 5 millimeters. (21 CFR 1020.33(g)(3))
11.1.7.2 If the x-ray production period is less than one-half second, the indication of x-ray production shall be actuated for at least one-half second. Indicators at or near the gantry shall be discernible from any point external to the patient opening where insertion of any part of the human body into the primary beam is possible. (second part of 21 CFR 1020.33(h)(1))
11.1.7.3 The deviation of indicated scan increment versus actual increment shall not exceed plus or minus 1 millimeter with any mass from 0 to 100 kilograms resting on the support device. The patient support device shall be incremented from a typical starting position to the maximum incremented distance or 30 centimeters, whichever is less, and then returned to the starting position. Measurement of actual versus indicated scan increment may be taken anywhere along this travel. (21 CFR 1020.33(i))
11.1.7.4 Premature termination of the x-ray exposure by the operator shall necessitate resetting of the CT conditions of operation prior to the initiation of another scan. (second part of 21 CFR 1020.33(f)(2)(ii)
11.2 CT Facility Design Requirements.
11.2.1 Aural Communication. Provision shall be made for two-way aural communication between the patient and the operator at the control panel.
11.2.2 Viewing Systems.
11.2.2.1 Windows, mirrors, closed-circuit television, or an equivalent shall be provided to permit continuous observation of the patient during irradiation and shall be so located that the operator can observe the patient from the control panel.
11.2.2.2 When the primary viewing system is by electronic means, an alternate viewing system (which may be electronic) shall be available for use in the event of failure of the primary viewing system.
11.3 CT Surveys, Performance Evaluations, Routine QC, and Operating Procedures.
11.3.1 Radiation Protection Surveys.
11.3.1.1 All CT x-ray systems installed after the effective date of these regulations shall have a radiation protection survey completed by, or under the direct supervision of, the QMP prior to clinical use. Existing systems not previously surveyed shall have a survey made by, or under the direct supervision of, a QMP within 12 months after the effective date of these regulations. In addition, such surveys shall be done after any change in the facility or equipment which might cause a significant increase in radiation hazard.
11.3.1.2 The registrant shall obtain a written report of the survey from the QMP (QE), and a copy of the report shall be made available to the Agency upon request.
11.3.2 System Performance Evaluations.
11.3.2.1 The annual testing of the CT x-ray system shall be performed by, or under the personal supervision of, a QMP who assumes the responsibility and signs the final performance evaluation report.
11.3.2.2 Evaluation standards and tolerances shall be established by the QMP (QE) and maintained by the facility. These standards and tolerances shall meet nationally recognized standards and tolerances for the CT x-ray system.
11.3.2.3 The evaluation of a CT x-ray system shall be performed after initial installation, and at least annually thereafter. In addition, the QMP shall complete an evaluation of the CT system prior to clinical use, after any change or replacement of components which, in the opinion of the QMP, could cause a change in the radiation output or image quality.
11.3.2.4 The evaluation shall include but not be limited to:
11.3.2.4.1 Geometric factors and alignment including:
11.3.2.4.1.1 Alignment light accuracy;
11.3.2.4.1.2 Table increment accuracy.
11.3.2.4.2 Image localization from scanned projection radiograph (localization image);
11.3.2.4.3 Radiation beam width;
11.3.2.4.4 Image quality including:
11.3.2.4.4.1 High-contrast (spatial) resolution;
11.3.2.4.4.2 Low-contrast resolution;
11.3.2.4.4.3 Image uniformity;
11.3.2.4.4.4 Noise;
11.3.2.4.4.5 Artifact evaluation.
11.3.2.4.5 CT number accuracy;
11.3.2.4.6 Image quality for acquisition workstation display devices;
11.3.2.4.7 A review of the results of the routine QC required under Part F subsection 11.1.3;
11.3.2.4.8 A safety evaluation of audible and visual signals, posting requirements;
11.3.2.4.9 Dosimetry.
11.3.2.5 The measurement of the radiation output of a CT x-ray system shall be performed with a calibrated dosimetry system. The calibration of such system shall be traceable to a national standard. The dosimetry system shall have been calibrated within the preceding 2 years.
11.3.3 Routine Quality Control. A routine QC program on the CT system shall:
11.3.3.1 Be developed by a QMP (QE) and include acceptable tolerances for points evaluated;
11.3.3.2 Incorporate the use of a water equivalent phantom. At a minimum, noise, CT number, and artifacts shall be evaluated.
11.3.3.3 Be completed at time intervals and under system conditions specified by the QMP (QE). The interval shall not to exceed 1 week.
11.3.3.4 Be documented and maintained for inspection by the Agency.
11.3.4 Operating Procedures.
11.3.4.1 The operator of the CT x-ray system shall meet the minimum operator requirements of these regulations and be specifically trained on the operational features of the unit by a manufacturer's applications specialist, and/or QMP.
11.3.4.2 The following information shall be readily available to the CT operator:
11.3.4.2.1 Instructions on performing routine QC, including the use of the CT phantom(s), a schedule of routine QC appropriate for the system, allowable variations set by the QMP (QE) for the indicated parameters, and the results of at least the most recent routine QC completed on the system; and
11.3.4.2.2 Scanning protocols established by the RPC, including instructions on reporting deviations.
11.3.4.3 If the QMP (QE) evaluation or routine QC of the CT x-ray system identifies that a system operating parameter has exceeded a tolerance established by the QMP (QE), use of the CT x-ray system on patients shall be limited to those uses permitted by established written instructions of the QMP (QE).
11.4 Radiation Protocol Committee (RPC). The registrant shall develop and maintain an RPC in accordance with the following:
11.4.1 Members of the RPC.
11.4.1.1 Members of the RPC shall include but not be limited to a:
11.4.1.1.1 Radiologist;
11.4.1.1.2 CT Technologist;
11.4.1.1.3 QMP or QE; and
11.4.1.1.4 Other individuals as deemed necessary by the registrant (e.g., Radiation Safety Officer, Chief Medical or Administrative Officer, Radiology Department Administrator/Manager).
11.4.2 If the registrant has more than one site with CT, they may establish a system-wide RPC.
11.4.3 Two or more registrants may form a cooperative RPC as long as each facility has a representative on the committee.
11.4.4 If the registrant has already established a radiation safety or Radiation Protocol Committee, the requirements of this subsection may be delegated to that committee if the members meet the requirements of Part F, subsection 11.5.1
11.4.5 Responsibilities of the RPC. The RPC shall:
11.4.5.1 Review existing CT protocols along with the evaluation and implementation of new and innovative technologies that can improve image quality and/or lower patient dose in comparison with the older protocol.
11.4.5.2 Review the capabilities of the individual CT scanner to ensure maximum performance is achieved.
11.4.5.3 Determine and review the protocols used frequently or could result in significant doses. This review shall include acquisition and reconstruction parameters, image quality, and radiation dose. At a minimum, the facility shall review the following clinical protocols, if performed, at least annually:
11.4.5.3.1 Pediatric Head;
11.4.5.3.2 Pediatric Abdomen;
11.4.5.3.3 Adult Head;
11.4.5.3.4 Adult Abdomen;
11.4.5.3.5 Adult Chest;
11.4.5.3.6 ) Brain Perfusion.
11.4.5.4 Establish and implement written protocols, or protocols documented in an electronic reporting system, that include but are not limited to the following:
11.4.5.4.1 A method to be used to monitor the CT radiation output.
11.4.5.4.2 A standardized protocol naming policy.
11.4.5.4.3 A DRL, notification value, and alert value for CT procedures reviewed in Part F, subsection 11.5.2.3. Notification and alert values may be applied by using trigger values in conformance with NEMA XR-29 or facility-established values and procedures as defined by the QMP.
11.4.5.4.4 Actions to be taken for cases when the dose alert value was exceeded which may include patient follow-up.
11.4.5.4.5 A process determining who has access and authority to make changes to the protocol management systems, including a method to prevent inadvertent or unauthorized modifications to a CT protocol.
11.4.5.5 If CT fluoroscopy is performed, the RPC shall establish and implement operating procedures and training designed to minimize patient and occupational radiation exposure.
11.4.5.6 Provide an annual report to the radiation safety committee or radiation safety officer, in the absence of a radiation safety committee,
11.4.5.7 At a minimum the RPC members in Part F, subsection 11.4.1.1 shall meet as often as necessary to conduct business but at intervals not to exceed 12 months.
11.4.6 Records.
11.4.6.1 A record of each RPC meeting shall be maintained. The record shall include the date, names of individuals in attendance, minutes of the meeting, and any action taken, for at least three years.
11.4.6.2 The registrant shall maintain a record of RPC policies and procedures.
11.4.6.3 The registrant shall maintain a record of radiation output information so the radiation dose may be estimated in accordance with established protocols (e.g., SSDE). The record shall include:
11.4.6.3.1 Patient identification;
11.4.6.3.2 Type and date of examination;
11.4.6.3.3 Identification of the CT system used; and
11.4.6.3.4 The dose values the CT system provides (e.g., CTDIvol, DLP, SSDE).
11.5 CT systems used in treatment planning. CT systems solely used for treatment planning in radiation oncology shall meet the requirements in Part X.10 of these regulations.
11.6 PET CT and SPECT CT Systems. CT systems solely used to calculate attenuation coefficients in nuclear medicine studies shall meet the requirements in Sections Part F, subsection 11.1 through Part F, subsection 11.4 unless otherwise exempted below:
11.6.1 Part F, subsection 11.1.1 (Accreditation)
11.6.2 In lieu of Part F, subsection 11.3.2, a QMP shall complete a performance evaluation on the CT system following nationally recognized guidelines at intervals not to exceed 12 months.
11.6.3 In lieu of Part F, subsection 11.3.3, routine QC checks shall be completed at intervals not to exceed 1 week. These checks shall be established and documented by a QMP following nationally recognized guidelines.
11.6.4 Part F, subsection 11.3.4.2.2 (RPC)
11.7 Veterinary CT Systems. CT systems, including CBCT systems, solely used in non-human imaging shall meet the requirements of Part F, subsection 11.3.1 (radiation protection surveys) and are otherwise exempt from the standards of Part F, subsection 11.0.
11.8 Cone Beam Computed Tomography Systems.
11.8.1 CBCT facilities shall meet Part F, Section 4.0, Part F, subsection 6.1 and 6.11, and Part F, subsection 11.1.2 through Part F, subsection 11.1.7, as applicable.
11.8.2 Beam alignment. The x-ray field in the plane of the image receptor shall not exceed beyond the edge of the image receptor by more than 2 percent of the SID, when the axis of the x-ray beam is perpendicular to the plane of the image receptor. In addition, the center of the x-ray field shall be aligned with the center of the image receptor to within 2 percent of the SID.
11.8.3 A performance evaluation shall be performed by, or under the direct supervision of, a QMP. The evaluation shall follow nationally recognized standards and tolerances. The evaluation shall be performed within 30 days of initial installation and least annually, and prior to clinical use, after any change or replacement of components which, in the opinion of the QMP, could cause a change in the radiation output or image quality. The facility shall maintain documentation of the established standards and tolerances and testing results for at least three years.
11.8.4 The registrant shall follow the QC recommendations provided by the CBCT manufacturer. In the absence of manufacturer provided QC recommendations, the registrant shall implement and document QC guidelines established by a QMP in accordance with nationally recognized guidelines.
11.8.5 The registrant or RPC, if established, shall implement and document a policy addressing deviations from established protocols.
11.8.6 The CBCT x-ray system shall only be operated by an individual who has been specifically trained in its operation.
11.8.7 The following information shall be readily available to the CBCT operator:
11.8.7.1 Instructions on performing routine QC, including the use of the CBCT phantom(s), a schedule of routine QC appropriate for the system, allowable variations set by the QMP (QE), if required, for the indicated parameters, and the results of at least the most recent routine QC completed on the system.
11.8.8 Exemption. A QMP performance evaluation on CBCT systems capable of operating at no greater than 100 kV or 20 mA shall be performed at least every two years.
11.8.9 Exemption. The registrant using fluoroscopy systems capable of CBCT shall meet Part F subsection 11.8, except Part F, subsection 11.1.2 through Part F, subsection 11.1.7 in Part F, subsection 11.8.1.
12.0 through 14.0 - Reserved
15.1 DXA systems shall be:
15.1.1 Certified by the manufacturer pursuant to the Medical Device Act and Subchapter C – Electronic Product Radiation Control (EPRC) of Chapter V of the Federal Food, Drug and Cosmetic Act;
15.1.2 Registered in accordance with Part B of these regulations; and
15.1.3 At a minimum, maintained and operated in accordance with the manufacturer’s specifications.
15.2 Operator Requirements. In addition to the minimum qualifications outlined in these regulations, operators shall complete training specific to patient positioning and the operation of the DXA system.
15.3 During the operation of any DXA system:
15.3.1 In the absence of a survey performed by or under the supervision of a QMP (QE) determining the minimum distance the operator may be from the patient and radiation source, the operator, ancillary personnel, and members of the general public shall be positioned at least two meters from the patient and DXA system during the examination, or alternatively, work shall be performed using protective lead aprons and/or a portable shield.
15.4 Reserved.
15.5 Quality Assurance. In addition to the applicable requirements in Part F, subsection 3.2.1, a facility performing DXA shall:
15.5.1 Conform to the DXA system manufacturer recommendations and recommendations of recognized professional societies such as the International Society for Clinical Densitometry or the American College of Radiology;
15.6 Records. The registrant shall keep the following records for a minimum of 3 years:
15.7 The maintenance and QC tests as prescribed by Part F, subsection 15.1.3 and Part F, subsection 15.5.
PART F
INFORMATION TO BE SUBMITTED BY PERSONS
PROPOSING TO CONDUCT HEALING ARTS SCREENING
Persons requesting that the Agency approve a healing arts screening program shall submit the following information for evaluation and approval:
a. Name and address of the applicant and, where applicable, the names and addresses of agents within this State;
b. Diseases or conditions for which the x ray examinations are to be used in diagnoses;
c. A description of the x ray examinations proposed in the screening program i.e., type and number of views;
d. Description of the population to be examined in the screening program, i.e., age range, sex, physical condition, and other appropriate information;
e. An evaluation of any known alternate methods not involving ionizing radiation that could achieve the goals of the screening program and why these methods are not used instead of the x ray examinations;
f. An evaluation by a QMP (QE) of the x ray system(s) to be used in the screening program. The evaluation shall include the following:
1. Documentation that such system(s) satisfy all applicable requirements of these regulations;
2. Measurement of appropriate patient exposures from the x-ray examinations to be performed;
g. A description of the x-ray quality control program;
h. A copy of the protocol information for the x ray examination procedures to be used;
i. The qualifications of each individual who will be operating the x ray system(s);
j. The qualifications of the individual who will be supervising the operators of the x ray system(s). The extent of supervision and the method of work performance evaluation shall be specified;
k. The name and address of the practitioner licensed in the state who will interpret the radiograph(s);
l. Procedures to be used in advising the individuals screened and their practitioner of the healing arts or health care provider of the results of the screening procedure and any further medical needs indicated;
m. Procedures for the retention or disposition of the radiographs and other records pertaining to the x ray examinations;
n. Frequency of screening of individuals; and
o. The duration of the screening program.
This part provides for the licensing of radioactive material, for purposes of protecting the public health and safety. No person shall receive, possess, use, transfer, sell, own or acquire radioactive material except as authorized in a specific or general license per the U.S. Nuclear Regulatory Commission (NRC), in accordance with Title 10 – Code of Federal Regulations. Primary radioactive material licensing and enforcement authority was transferred to the NRC in 2007, pursuant to the Federal Energy Policy Act of 2005. However, radioactive material facilities must be registered with the State of Delaware in accordance with Part B of these regulations.
RADIATION SAFETY REQUIREMENTS FOR NON-HEALING ARTS RADIATION GENERATING DEVICES (RGD)
This Part provides special requirements for non-healing arts radiation generating devices (RGDs) operating between 5 kiloelectron volts (keV) and 1 million electron volts (MeV). For machines operating at energies greater than 1 MeV, see Part I, (Radiation Safety Requirements for Particle Accelerators) of these regulations.
2.1 In addition to the requirements of this Part, all registrants are subject to the requirements of Parts A, B, D, and J of these regulations. This Part does not pertain to radiation safety requirements for x-ray equipment that is explicitly covered in other sections of these regulations (e.g., Diagnostic Machines (Part F), Particle Accelerators (Part I), Therapy Machines (Part X) and Radiation Safety Requirements for Industrial Radiographic Operations (Part E)).
2.2 Radiography that meets the definition of "cabinet radiography" (Part H, Section 4.0) shall be regulated under this Part. This includes certified cabinet x-ray systems.
2.3 Radiography that occurs in a "shielded room" as defined in Part H, Section 4.0 shall be regulated under this Part.
2.4 Industrial radiography that is open-beam, and not in a shielded room and not otherwise listed here, shall be regulated under Part E (Radiation Safety Requirements for Industrial Radiographic Operations) of these regulations.
RGDs are a broad class of equipment that generate x-rays or particle radiation having energies between 5 keV and 1 MeV, and not intended for medical use on humans. If applicable, all RGDs shall comply with FDA performance standards as defined in Title 21 Code of Federal Regulations, parts 1010 thru 1050. Examples of RGDs include, but are not limited to: open and closed analytical x-ray equipment (table top and hand-held), x-ray gauges, cabinet x-ray radiography, security screening units, quality control application devices, ion implantation devices, electron beam welders, non-human use x-ray fluoroscopy, and x-ray irradiators. The intent here is not to define safety parameters by what type of work the x-ray unit performs (analytical, gauge, radiography, etc.), but to classify by hazard (open-beam versus closed-beam) or dose rate. All other non-enclosed beam industrial radiography shall be regulated under Part E of these Regulations (Radiation Safety Requirements for Industrial Radiographic Operations).
As used in this Part, the following definitions apply:
"Accessible surface" means the external or outside surface of the enclosure or housing provided by the manufacturer. This includes the high-voltage generator, doors, access panels, latches, control knobs, and other permanently mounted hardware and including the plane across the exterior edge of any opening.
"Analytical x-ray equipment" means equipment that generates (by electronic means) and uses ionizing radiation for the purpose of examining the microstructure of materials, i.e. diffraction and spectroscopy (including fluorescence).
“Annual” means approximately every 12 months and not to exceed 14 months.
"Baggage unit". See "Security Screening Unit".
"Beam-port" means an opening on the x-ray apparatus designed to emit a primary beam. This does not include openings on baggage units.
"Cabinet radiography" means industrial radiography using radiation machines not subject to FDA performance standard for cabinet x-ray systems, in an enclosed, interlocked cabinet in which the portion of a material being irradiated is contained, and in which:
i. The radiation machine will not operate unless all openings are closed with interlocks activated;
ii. The cabinet is shielded such that every location on the exterior meets the conditions for an unrestricted area as defined in Part D of these regulations; and
iii. The cabinet is constructed or arranged as to exclude the entrance of any part of the body of an individual during irradiation.
"Cabinet x-ray system" means an x-ray system with the x-ray tube installed in an enclosure which, independently of existing architectural structures except the floor on which it may be placed, is intended to contain at least that portion of a material being irradiated, provide radiation attenuation, and exclude personnel from its interior during generation of x radiation. An x-ray tube used within a shielded part of a building, or x-ray equipment which may temporarily or occasionally incorporate portable shielding is not a cabinet x-ray system.
"Cathode ray tube" means any device used to accelerate electrons for demonstration or research purposes, except where such cathode ray tube is incorporated into a television or display monitor that is subject to, and has met applicable federal radiation safety performance standards in 21 CFR 1010 and 1020.10.
"Certifiable cabinet x-ray system" means an existing uncertified RGD that has been modified to meet the certification requirements specified in 21 CFR 1020.40.
"Certified cabinet x-ray system" means a RGD certified by the manufacturer in accordance with 21 CFR 1010.2 as being manufactured and assembled pursuant to the provisions of applicable federal radiation safety performance standards in 21 CFR 1010 and 1020.40.
"Closed-beam x-ray equipment" means a system in which the beam path cannot be entered by any part of the body during normal operation.
"Cold-cathode gas discharge tube" means an electronic device in which electron flow is produced and sustained by ionization of contained gas atoms and ion bombardment of the cathode.
"Collimator" means a device for restricting the useful radiation in one or more directions.
“Cone Beam CT” (CBCT) means a method of computed tomography that creates a three-dimensional cone-shaped beam that circles the object once to generate an image.
"Control panel" means a device containing means for regulation and activation of a RGD or for the preselection and indications of operating factors.
"Emergency procedure" means the written pre-planned steps to be taken in the event of actual or suspected exposure of an individual in excess of administrative or regulatory limits. This procedure shall include the names and telephone numbers of individuals to be contacted as well as directives for processing the film badge or other personnel monitoring devices.
"Fail-safe design" means a design in which all realistically anticipated failures of indicators or safety components result in a condition in which individuals are safe from exposure to radiation. For example, if a light indicating “X-RAY ON” fails, the production of x-rays shall be prevented, or if a shutter status indicator fails, the shutter shall close.
"General-use system" means a personnel screening system that delivers an effective dose equal to or less than 0.25 microsieverts (uSv), or 25 microRem (urem) per screening. Given proper justification and certain restrictions, general-use systems may be operated without specific controls that would limit the number of individuals scanned or the number of scans per individual in a year.
"Hand-held x-ray system" means a portable instrument that is designed to operate when held in the hand, e.g., hand-held XRF analytical devices.
"Industrial radiography" means an examination of the structure of materials by nondestructive methods utilizing ionizing radiation to make radiographic images.
"Interlock" means a device or engineered system that precludes access to an area of radiation hazard either by preventing entry or by automatically removing or inactivating the hazard.
"Leakage radiation" means all radiation coming from within the source housing, except the useful beam.
"Limited-use system" means a personnel screening system that is capable of delivering an effective dose greater than 0.25 microsieverts (uSv), or 25 microRem (urem) per screening but cannot exceed an effective dose of 10 microsieverts (uSv), or 1 microRem (urem) per screening. Limited-use systems require additional controls and documentation to ensure that annual individual dose limits required by Part H, subsection 11.5 are not exceeded.
"Local components" means parts of a RGD x-ray system and include areas that are struck by x¬-rays such as radiation source housings, beam port and shutter assemblies, collimators, sample holders, cameras, goniometers, detectors, and shielding, but do not include power supplies, transformers, amplifiers, readout devices, and control panels.
"Mobile equipment" See "Radiation generating device."
"Normal operating procedures" mean step-by-step instructions necessary to accomplish the task. These procedures may include sample insertion and manipulation, equipment alignment, routine maintenance by the registrant, and data recording procedures, which are related to radiation safety.
"Open-beam x-ray equipment" means an open-beam x-ray system in which the beam path could be entered by any part of the body at any time.
"Personnel security screening system" means any x-ray equipment used on humans for security evaluation.
"Portable equipment" See "Radiation generating device."
"Primary beam" means the ionizing radiation coming directly from the radiation source through a beam port into the volume defined by the collimation system.
"Qualified expert" means an individual who has satisfactorily fulfilled the training and experience requirements consistent with achieving a level of competency sufficient to function effectively in the position for which registration is sought, in accordance with 4465 Part B. Such individuals must demonstrate to the satisfaction of the Agency their qualifications, for example, individuals certified in the appropriate field by the American Board of Radiology, or the American Board of Health Physics, or the American Board of Medical Physics, or those having equivalent qualifications.
"Radiation generating device (or RGD)" means any system, device, subsystem, or component thereof, which may generate x-rays or particle radiation between 5 keV and 1 MeV, and not intended for healing arts use for humans or animals. A RGD may be fixed or portable, such as:
i. Mobile means RGD equipment mounted on a permanent base with wheels and/or casters for moving while completely assembled;
ii. Portable means RGD equipment designed to be hand-carried;
iii. Stationary means RGD equipment that is installed or placed in a fixed location; or
iv. Transportable means RGD equipment to be installed in a vehicle or that may be readily disassembled for transport or use in a vehicle.
"Radiation Safety Officer (RSO)" means an individual as defined in Part A, Appendix C of these regulations.
"Radiation source (or x-ray tube) housing" means that portion of an x-ray system which contains the x-ray tube and/or secondary target. Often the housing contains radiation shielding material or inherently provides shielding.
"Radiograph" means a permanent film or digital image produced on a sensitive surface by a form of radiation other than direct visible light or other forms of non-ionizing radiation.
"Radiography" is the process of creating radiographic images.
"Safety device" means a device, interlock or system that prevents the entry of any portion of an individual’s body into the primary x-ray beam or that causes the beam to shut off upon entry into its path.
"Scattered radiation" means radiation that has been deviated in direction and / or energy by passing through matter.
"Security screening unit" means a non-human use open-beam or cabinet x-ray system with accessible openings designed for the detection of weapons, bombs, or contraband concealed in baggage, mail, packages or other commodities or structure.
"Shielded room" means a room housing a RGD where, with the RGD at maximum techniques, the exterior room environs meets the unrestricted area limits of 0.02 millisieverts (mSv), or 2 milliRem (mrem) in any one hour and 1 millisieverts (mSv), or 100 milliRem (mRem) in a year at 30 cm from the barrier. A shielded room does not include a RGD which meet the definition of cabinet x-ray systems.
"Shutter" means a moveable device used to block the useful (or primary) beam emitted from an x-ray tube assembly.
"Source" means the point of origin of the radiation, for example, the focal spot of an x-ray tube.
"Stationary equipment". See "Radiation generating device."
"Stray radiation" means the sum of leakage and scatter radiation.
"Warning device" means a visible or audible signal that warns individuals of a potential radiation hazard.
"X-ray gauge" means an x-ray producing device designed and manufactured for the purpose of detecting, measuring, gauging, or controlling thickness, density, level, or interface location.
"X-ray generator" means that portion of an x-ray system which provides the accelerating high voltage and current for the x-ray tube.
5.1 Unless utilized in a dedicated location, hand-held RGDs are exempt from the requirements of Section 6.0 Posting of the General Regulatory Provisions of this Part.
5.2 The following machines and equipment are exempt from these regulations:
5.2.1 Domestic television receivers, computer monitors, and electron microscopes, providing the exposure rate at 5 centimeters from any outer surface is less than 0.005 millisieverts (mSv), or 0.5 millirem (mRem) per hour.
5.2.2 Cold-cathode gas discharge tubes, providing the exposure rates shall not exceed 0.1 millisieverts (mSv), or 10 milliRem (mRem) per hour at a distance of thirty (30) centimeters from any point on the external surface of the tube.
5.2.3 Other electrical equipment that produces radiation incidental to its operation for other purposes, providing the dose rate to the whole body at the point of nearest approach to such equipment when any external shielding not integral to the equipment is removed does not exceed 0.25 microsieverts (uSv), or 25 microRem (urem) per year. The production testing or factory servicing for such equipment shall not be exempt.
5.2.4 Equipment described in this subsection shall not be exempt if it is used or handled in such a manner that any individual might receive a dose of radiation in excess of the limits specified in Part D of these regulations.
6.1 Unless otherwise provided in this Part, this Section applies to all Radiation Generating Devices (RGDs). Certified and Certifiable Cabinet X-ray Systems as defined in this Part shall also meet the requirements of 21 CFR 1020.40.
6.2 Warning Devices.
6.2.1 Warning devices shall be labeled so that their purpose is easily identified.
6.2.2 An easily visible warning device light labeled with the words "X-RAY ON," or words having a similar intent, shall be located a) near any switch that energizes an x-ray tube or b) in a conspicuous location near the radiation source housing and radiation beam and visible from all instrument access areas and shall be illuminated only when the tube is energized. This warning light shall be of a fail-safe design.
6.3 Labeling.
6.3.1 All RGD equipment shall be labeled with a readily visible and discernible sign or signs bearing the radiation symbol (defined in Part D.901 of these regulations) and the words: "CAUTION RADIATION - THIS EQUIPMENT PRODUCES RADIATION WHEN ENERGIZED," or words having a similar intent, near any switch that energizes an x-ray tube.
6.3.2 For RGDs with designed openings, for object entries (such as baggage units), the following shall be posted at or near each opening: "CAUTION - X-RAY HAZARD: DO NOT INSERT ANY PART OF THE BODY WHEN SYSTEM IS ENERGIZED", or words having similar intent.
6.4 Radiation Source Housing. Each x-ray tube housing shall be subject to the following requirements:
6.4.1 Interlock. When the x-ray tube housing is the primary shielding for the x-ray tube, and is intended to be opened for normal use or maintenance, the housing shall be equipped with an interlock that shuts off the high voltage to the x-ray tube if the housing is opened; and
6.4.2 Radiation Emission Limit. Each x-ray tube housing shall be so constructed that, with all shutters closed, the leakage radiation measured at a distance of 5 centimeters from the x-ray tube housing surface does not exceed 0.025 millisieverts (mSv), or 2.5 milliRem (mRem) per hour. This limit shall be met at the maximum tube rating. For closed-beam systems, this requirement can be met by complying with Part H, subsection 7.5, Radiation Emission Limit. For a RGD in a shielded room, this limit can be met by measuring from any accessible surface outside the room housing the RGD. For hand-held, open-beam RGDs, this requirement can be met by complying with the limits in Part H, subsection 9.4, Radiation Emission Limit.
6.5 Generator Cabinet or High Voltage Source Radiation Emission Limits. Each x-ray generator or high-voltage source shall be supplied with a protective cabinet which limits leakage radiation to 0.025 millisieverts (mSv), or 2.5 milliRem (mRem) per hour at a distance of 5 centimeters measured at the nearest accessible surface. For closed-beam systems, this requirement can be met by complying with Part H, subsection 7.5, Radiation Emission Limit. For a RGD in a shielded room with the high-voltage generator also inside the shielded room, this limit can be met by measuring from any accessible surface outside the room housing the RGD. For hand-held, open-beam RGDs, this requirement can be met by complying with the limits in Part H, subsection 9.4, Radiation Emission Limit.
6.6 Surveys.
6.6.1 Radiation surveys of all RGDs shall be sufficient to show compliance with radiation emission requirements of this Part, and as required by Part D.201 (Occupational Dose Limits for Adults) and Part D.301 (Dose Limits for Individual Members of the Public) of these regulations. The radiation surveys shall be sufficient to evaluate the magnitude and extent of radiation emissions and the potential radiological hazards that could be present. At a minimum, surveys shall be performed:
6.6.1.1 Upon installation of the equipment, and at least annually thereafter;
6.6.1.2 Following any change in the initial arrangement, number, or type of local components in the system;
6.6.1.3 Following any maintenance requiring the disassembly, removal, or repair of a local component in the system;
6.6.1.4 During the performance of maintenance, calibration and other procedures if the procedures require the presence of a primary x-ray beam while any local component in the system is disassembled or removed;
6.6.1.5 Post bypass of a safety device or interlock as required by Part H, subsection 6.8.2;
6.6.1.6 Any time a visual inspection of the local components in the system reveals an abnormal condition;
6.6.1.7 Whenever a personnel monitoring device shows a significant increase over previous monitoring period or readings are approaching the limits specified in Part D.201 (Occupational Dose Limits for Adults) of these regulations.
6.6.2 The registrant shall have access to sufficiently calibrated, appropriate and operable radiation survey instruments to make physical radiation surveys as required by this Part. The instruments shall be capable of detecting and measuring the types and levels of radiation involved (including primary, scattered, and leakage radiation).
6.6.3 The registrant shall assure the maintenance and calibration of all monitoring and survey instruments per Part D.501 of these regulations.
6.6.4 Radiation survey measurements shall not be required if a registrant can otherwise demonstrate compliance with the requirements of this Part to the satisfaction of the Agency.
6.6.5 Posting. Each area or room containing an RGD where an individual may receive 0.02 millisieverts (mSv), or 2 milliRem (mRem) in any one hour or 1 millisieverts (mSv), or 100 milliRem (mRem) per year shall be conspicuously posted with a sign or signs bearing the radiation symbol (as defined in Part D.901 of these regulations) and the words "CAUTION - X-RAY EQUIPMENT," "CAUTION - RADIATION GENERATING DEVICE" or words having a similar intent.
6.6.6 Security. RGDs shall be secured in such a way as to be accessible to, and operated by only authorized and trained personnel.
6.6.7 Operating Requirements.
6.6.7.1 Procedures. Normal operating procedures shall be written and available to all RGD workers. No individual shall be permitted to operate a RGD in any manner other than that specified in the procedures unless such individual has obtained the written approval of the radiation safety officer.
6.6.7.2 Bypassing.
6.6.7.2.1 No individual shall bypass a safety device, interlock, or remove shielding unless such individual has obtained the written approval of the radiation safety officer. Such approval shall be for a specified period of time.
6.6.7.2.2 When a safety device or interlock has been bypassed, a readily discernible sign bearing the words "SAFETY DEVICE NOT WORKING," or words having a similar intent, shall be placed on the radiation source housing and at the control switch.
6.6.7.2.3 A record of any bypass of a safety device or interlock shall be maintained; the record shall contain such information as the date the alteration was made, type of alteration, length of time the unit remained in the altered condition, post bypass survey and signed by the RSO, individual who made the alteration, and the individual who restored the unit to original condition.
6.6.7.3 Control Panel.
6.6.7.3.1 The RGD can only be activated from a control panel.
6.6.7.3.2 All indicators and controls that control the primary beam shall be identifiable and discernible through the use of labels, symbols, software displays or the equivalent.
6.6.7.4 Interlocks.
6.6.7.4.1 An interlock shall not be used to de-activate the x-ray tube or RGD, except in an emergency or during testing of the interlock system.
6.6.7.4.2 After triggering any interlock, it shall be possible to reset the RGD to full operation only from a control panel.
6.6.7.4.3 All interlocks shall be of a fail-safe design.
6.6.7.5 Multiple Sources. If more than one x-ray tube assembly(s) or focal spot can be operated sequentially or simultaneously from a control panel, visual indicators shall identify which tube assembly(s) or focal spot has been selected. The selectors shall be identified as to their function. If a letter or number is used, a reference card or table explaining the code shall be affixed to the control panel.
6.6.8 Repair or Modification of X-Ray Tube or RGD Systems. Only trained internal personnel authorized by the registrant's senior management, or state-registered Radiation Service Providers shall be permitted to install, repair, or make modifications to the RGD. No operation involving removal of covers, shielding materials or tube housings or modifications to shutters, collimators, or beam stops shall be performed without ascertaining that the tube is off and will remain off until safe conditions have been restored. The main power switch with a lock-out / tag-out, rather than interlocks, shall be used for routine shutdown in preparation for repairs. It is the responsibility of the registrant to assure that qualified personnel install, repair, or make modifications to the RGD.
6.6.9 Testing of Safety Devices.
6.6.9.1 Tests of all safety devices, such as interlocks, shutters, warning lights, and required emergency shut-off switches shall be conducted at intervals not to exceed 12 months on all operable RGDs.
6.6.9.2 If any safety device fails during testing, the RGD shall be removed from service until the safety device failure is corrected or proper temporary administrative controls established and approved in writing by the RSO.
6.6.9.3 Records of safety device tests, check dates, findings and corrective actions shall be available for inspection and maintained for 5 years.
6.6.9.4 Records shall include the date of the test, a list of the safety devices tested, survey instrument information, calibration date, the results of the test, the name of the person performing the tests and corrective actions taken for safety devices that fail the required test.
6.6.9.5 Testing of safety devices may be deferred if the unit and/or installation is clearly marked and kept out of service; units and/or installations brought back into service after exceeding the 12 month interval shall be tested prior to use.
6.6.9.6 If testing of a safety device cannot be performed due to manufacturer design, the registrant shall document that the safety device will not be tested and specifically why the safety device cannot be tested.
6.6.10 Instruction and Training. The registrant shall document the scope of training required for the RGD they possess in accordance with this section. No individual shall be permitted to operate or maintain an RGD, or enter a shielded room without appropriate instruction and training. Records shall be maintained onsite of all required training and instruction, and made available for review by the Agency. Each such individual shall receive instruction in and demonstrated competence as to:
6.6.10.1 Types of radiation and identification of radiation hazards associated with the use of the RGD and associated equipment and precautions or measures necessary to minimize radiation exposure;
6.6.10.2 Significance of the various radiation warning, safety devices, and interlocks incorporated into the equipment, or the reasons they have not been installed on certain pieces of equipment and the extra precautions required in such cases;
6.6.10.3 Commensurate with potential hazards of use, biological effects of radiation, radiation risks, and recognition of symptoms of an acute localized exposure;
6.6.10.4 Normal operating procedures for each type of RGD and associated equipment, including having received hands-on training, and procedures to prevent unauthorized use;
6.6.10.5 Procedures for reporting an actual or suspected accidental exposure or other radiation safety concerns, such as any unusual occurrence or malfunction that may involve exposure to radiation; and
6.6.10.6 Performing surveys where applicable.
6.6.11 Radiation Protection Responsibility.
6.6.11.1 The registrant's senior management shall make the ultimate decision to use any Radiation Generating Devices (RGD), and be ultimately responsible for radiation safety.
6.6.11.2 The registrant's senior management shall designate an individual responsible for radiation safety, or a Radiation Safety Officer (RSO). This individual shall have direct access to senior management for radiation safety issues. This individual shall have training and experience commensurate with the scope of the radiation safety program to carry out the responsibilities as indicated below.
6.6.11.2.1 Establishing and overseeing operating and safety procedures that maintain radiation exposures as low as reasonably achievable (ALARA), and to review them periodically to ensure that the procedures are current and conform with these regulations;
6.6.11.2.2 Ensuring that individual monitoring devices are properly used by occupationally exposed personnel as required by the regulations, that records are kept of the monitoring results, and that timely notifications are made as required by Part D;
6.6.11.2.3 Investigating and reporting to the agency each known or suspected case of radiation exposure to an individual or radiation level detected in excess of limits established by these regulations and each theft or loss of source(s) of radiation, determining the cause, and taking steps to prevent its recurrence;
6.6.11.2.4 Having a thorough knowledge of management policies, administrative procedures and records of the registration permit-holder and keeping management informed on a periodic basis of the performance of the registrant's radiation protection program, if applicable;
6.6.11.2.5 Assuming control and having the authority to institute corrective actions including shutdown of operations when necessary in emergency situations or unsafe conditions;
6.6.11.2.6 Maintaining records as required by these regulations; and
6.6.11.2.7 Ensuring that personnel are adequately trained and complying with these regulations, the conditions of the certificate of registration, and the operating and safety procedures of the registered permit-holder.
6.6.11.2.8 Ensuring that safety devices, interlocks, warning signals, labels, postings, and signs are functioning and located where required.
7.1 In addition to the requirements of Part H, Section 6.0, the following applies to all closed-beam x-ray RGDs.
7.2 System Enclosure. The radiation source, sample or object, detector, and analyzing crystal (if used) shall be enclosed in a chamber or coupled chambers that cannot be entered by any part of the body during normal operation.
7.3 Interlocks. All doors and panels accessing the RGDs shall be interlocked. The interlocks required by this section shall be of a fail-safe design.
7.4 Interlock Functions. The system enclosure, sample chamber, etc. closure shall be interlocked with the x-ray tube high voltage supply and/or a shutter in the primary beam so that no x-ray beam can enter the sample or object chamber while it is open unless the interlock has been conspicuously and deliberately defeated. The interlock required by this section shall be of fail-safe design or adequate administrative controls shall be exercised to ensure operations will not continue without a proper functioning interlock.
7.5 Radiation Emission Limit. The radiation emission for all closed beam RGDs shall not exceed a dose rate of 0.005 millisieverts (mSv), or 0.5 milliRem (mRem) in one hour at five centimeters outside any accessible surface.
7.6 Security Screening Units. Security screening units shall be provided with means to ensure operator presence at the control area in a position which permits surveillance of the openings and doors during generation of x-radiation.
7.7 During an exposure or preset succession of exposures of one-half second or greater duration, the means provided shall enable the operator to terminate the exposure or preset succession of exposures at any time.
7.8 During an exposure or preset succession of exposures of less than one-half second duration, the means provided may allow completion of the exposure in progress but shall enable the operator to prevent additional exposures.
8.1 In addition to the requirements in Section H.6, the following requirements apply to all open beam RGDs not otherwise addressed in this Part.
8.2 Safety Device.
8.2.1 The registrant shall document their justification of the use of open-beam instead of closed-beam systems.
8.2.2 If the registrant needs to use an open-beam system, the registrant shall consider a safety device which prevents the entry of any portion of the operator's body into the path of the primary beam or which causes the primary beam to be shut off upon entry into its path.
8.2.3 If the registrant's use of the open-beam RGD does not permit the use of a safety device to prevent direct body exposure, the registrant shall maintain a written record of a description of the various safety devices that have been evaluated and reasons for why these devices cannot be used. These records shall be available onsite for inspection.
8.2.4 In lieu of the safety device described in section Part H, subsection 8.2.2 above, the registrant shall employ alternative methods (such as policies and procedures) to minimize the possibility of unnecessary exposure. These alternative methods shall be documented. The documentation shall include information about the absence of safety devices. This documentation shall be available for inspection as long as these methods are employed, plus an additional 5 years.
8.2.5 For portable open-beam RGDs that are manufactured to be used hand-held, or potentially used as a hand-held, without such safety devices, this safety device requirement may be met by complying with all the requirements in Part H, Section 9.0, Additional Requirements for Open-beam, Hand-held RGDs prior to use.
8.3 X-ray On Status. For open beam equipment, RGDs shall be provided with a readily discernible and active indication of:
8.3.1 X-ray tube "on-off" status located near the radiation source housing. The warning lights as required by Part H, subsection 6.2.2 can meet this requirement if the warning lights are readily discernible and viewable by anyone near the primary beam;
8.3.2 Shutter "open-closed" status located at the control panel and near each beam port on the radiation source housing, if the primary beam is controlled with a shutter. The shutter status device shall be clearly labeled as to the meaning of the status device (i.e., whether the shutter is open or closed). The status light at the control panel can meet the requirement for the status light at the beam port if the status light at the control panel is readily discernible and viewable by anyone near the primary beam; and
8.3.3 The x-ray tube "on-off" status indicator and the shutter "open-closed" status indicators shall be of a fail-safe design.
8.4 Labeling. Each unit will be labeled at or near the x-ray exit beam port to identify the location of the beam with the words, "CAUTION - X-RAY BEAM", "CAUTION - HIGH INTENSITY X-RAY BEAM", or words having a similar intent.
8.5 Beam Ports. Unused beam ports on radiation source housings shall be secured in the closed position in a manner which will prevent inadvertent opening.
8.6 Shutters. On open-beam RGD configurations that are designed to accommodate interchangeable components, each beam port on the radiation source housing shall be equipped with a shutter that cannot be opened unless a collimator or a component coupling has been connected to the beam port.
8.7 Radiation Emission Limits. The local components of an open-beam RGD shall be located and arranged and shall include sufficient shielding or access control such that no radiation emissions exist (exclusive of the primary beam) in any area surrounding the local component group which could result in a dose to an individual present therein in excess of the dose limits as outlined in Part D. 1301 (Dose Limits for Individual Members of the Public) of these regulations. These emissions shall be met at any specified tube rating.
8.8 Primary Beam Attenuation. In cases where the primary x-ray beam is not intercepted by the detector device under all conditions of operation, protective measures shall be provided, such as auxiliary shielding or administrative procedures, to avoid exposure to any individual from the transmitted primary x-ray beam.
8.9 Operator Attendance. The operator shall be in immediate attendance at all times when the equipment is in operation except when the area is locked or the equipment is secured to protect against unauthorized or accidental entry.
8.10 Control of Access. If the RGD is not in a restricted area (as defined in Part A of these regulations), the operator shall be able to control access to the RGD at all times during operation. If the RGD is not in a restricted area (as defined in Part A) and the RGD is capable of creating a radiation area or a high radiation area (as defined Part A), the operator shall be able to control access to the RGD at all times during operation, and:
8.10.1 Radiation areas shall be conspicuously identified. The radiation source shall be within a conspicuous perimeter (e.g., rope, tape, or other barrier) that identifies the area in which the dose equivalent exceeds 0.05 millisieverts (mSv), or 5 milliRem (mRem) in one hour. The area described by the temporary barricade shall be suitably posted with "CAUTION -RADIATION AREA" signs. The operator shall ensure that no one is inside or enters the radiation area during operation of the RGD;
8.10.2 High radiation areas shall be conspicuously identified. The radiation source shall be within a conspicuous perimeter (e.g., rope, tape, or other barrier) that identifies the area in which the dose equivalent exceeds 1 millisieverts (mSv), or 100 milliRem (mRem) in one hour. The area described by the temporary barricade shall be suitably posted with "CAUTION - HIGH RADIATION AREA" signs. The operator shall ensure that no one is inside or enters the high radiation area during operation of the RGD;
8.10.3 The operator shall perform a visual check of the controlled area to ensure it is free of all unauthorized personnel immediately prior to activating or exposing the radiation source;
8.10.4 Surveillance of the exposure area shall be maintained during operation, either by visual or by other reliable means to ensure that no person enters the area;
8.10.5 With the exception of hand-held x-ray systems, following the conclusion of an exposure, the operator shall use a suitable calibrated and operable radiation detection instrument to verify that the radiation source is in its fully shielded condition or that the x-ray tube has been de-energized; or,
8.10.6 A personal alarming dose rate meter may be worn to approach the work area if the device is appropriately designed and calibrated for the type of x-ray emitted (i.e., pulse or continuous), set at an appropriate level to detect the presence of the source, for example 0.02 millisieverts (mSv), or 2 milliRem (mRem) per hour, and has been source-checked prior to use. The radiation in the work area must be reasonably uniform so that the device responds to radiation exposure to any part of the body. It may not be used to measure radiation levels, nor may it be used to indicate the presence of the source for potential non-uniform exposure, such as may occur during machine maintenance or work in a RGD target area;
8.10.7 Measurement of radiation levels for a radiation survey shall be performed using an appropriate calibrated radiation survey meter (see Part H, subsections 6.6.1 and 6.6.2). A radiation survey meter shall also be used when there is potential for non-uniform exposure to personnel, such as may occur during machine maintenance or work in a RGD target area;
8.10.8 During the initial exposure, the radiation levels shall be measured around the perimeter of the controlled area. The perimeter shall be adjusted accordingly to meet the access control requirement for radiation areas or high radiation areas; and;
8.10.9 The survey around the perimeter shall be made for each new operating condition and the perimeter adjusted accordingly. The area of operation shall be monitored periodically if radiation levels are variable.
8.10.10 Instruction and Training. In addition to the requirements in Part H, subsection 6.11, no individual shall be permitted to operate or maintain an open-beam RGD unless such individual has received more specific and detailed instruction in and documented, demonstrated competence as to:
8.10.10.1 Sources and magnitude of common radiation exposure;
8.10.10.2 Units of radiation measurement;
8.10.10.3 Radiation protection concepts of time, distance, shielding, and ALARA;
8.10.10.4 Procedures and rights of a declared pregnancy;
8.10.10.5 Regulatory requirements and area postings;
8.10.10.6 Worker, embryo/fetus, and public dose limits;
8.10.10.7 Proper use of survey instruments and dosimetry; and
8.10.10.8 The policies and procedures required by H.8a.
8.10.11 Personnel Monitoring. In addition to the requirements of Part D.201 of these regulations (Occupational Dose Limits for Adults), extremity dosimetry shall be provided and used by:
8.10.11.1 Personnel working with or routinely working near and having potential for exposure to, the primary beam of an open-beam RGD; and
8.10.11.2 Personnel maintaining RGDs if the maintenance procedures require the presence of a primary radiation beam when any local component in the RGD is disassembled or removed.
9.1 In addition to the requirements in Part H, Sections 6.0 and 8.0, the following requirements in this Section apply to open-beam, hand-held RGDs.
9.2 Procedures. All registrants possessing open-beam, hand-held RGDs shall have available for review to the Agency operating policies and procedures that contain measures to insure that:
9.2.1 Radiation protection is provided equivalent to that afforded in Part D.301 of these regulations (Dose Limits for Individual Members of the Public);
9.2.2 Radiation protection is provided equivalent to that afforded in Part H, subsection 8.8 (Primary Beam Attenuation);
9.2.3 The operator will not hold the object or target during operation of the RGD and that the operator's hands will not approach the primary beam;
9.2.4 The operator will not aim the primary beam at him/herself or at any individual during operation of the RGD; and
9.2.5 Operator radiation exposure is as low as reasonably achievable (ALARA), for example, by use of ancillary equipment that will reduce exposure.
9.3 Training. In addition to the training requirements of Part H, subsection 6.11 and Part H, subsection 8.10.10 above, the registrant shall provide training for all users and operators on the subjects in Part H, subsection 9.2. Records shall be maintained of all user and operator training.
9.4 Radiation Emission Limit. For hand-held RGDs, the limits of Part H, subsection 6.4.2 (Radiation Source Housing Radiation Emission Limits) and Part H, subsection 6.5 (Generator Cabinet or High Voltage Source Radiation Emission Limits), excluding the primary beam, shall be met if the radiation emission at any accessible surface of the RGD does not exceed 0.025 millisieverts (mSv), or 2.5 milliRem (mRem) per hour at 5 cm.
9.5 Extremity Monitoring. For the purposes of the requirements in Part H, subsection 8.10.11 (extremity monitoring), operators of hand-held RGDs shall be considered as working near the primary beam. Extremity dosimeters are required if dose may exceed 10% of the annual occupational dose limit.
10.1 For RGDs that do not meet the limits of Part D.301 (Dose Limits to Individual Members of the Public), the RGD can be maintained inside a shielded room such that the exterior of the room meets the limits of Part D.301 of these regulations (Dose Limits to Individual Members of the Public) when the RGD is activated. RGDs in a shielded room shall be required to meet only the requirements of Part H, Section 6.0 (General Requirements) and the following.
10.2 Posting. The door to the room containing the RGD shall be posted "CAUTION - RADIATION AREA", or "CAUTION - HIGH RADIATION AREA", or "GRAVE DANGER - VERY HIGH RADIATION AREA", as required by Part D of these regulations.
10.3 Entrance Interlocks. All entrances into the shielded room shall be provided with interlocks. After an interlock has been interrupted, broken, or tripped, it shall be possible to cause x-rays to be produced again only from the control panel. Interlocks shall not be used to shut off the x-ray equipment except in an emergency or during testing.
10.4 Entrance Warning Devices. All entrances into the shielded room shall be provided with a conspicuously visible warning device, which need not be flashing or rotating but which operates only when radiation is being produced. The warning device shall be labeled in accordance with Part H, subsection 6.2.
10.5 Room Warning Lights. The interior of the shielded room shall be provided with flashing or rotating warning lights that operate when, and only when, radiation is being produced. These lights shall be positioned so that they can be observed from any position or orientation within the room. The lights shall be posted indicating the meaning of the warning signal and instructions on what to do; the posting shall be legible, conspicuous, and accessible to view by personnel both inside and outside the room.
10.6 Audible Room Warning Device. An audible warning signal within the room shall be actuated for at least ten (10) seconds immediately prior to the first initiation of radiation after the closing of any opening that can admit personnel. The registrant shall post the meaning of the warning signal and instructions on what to do; the posting shall be legible, conspicuous, and accessible to view by personnel within the room.
10.7 Emergency Shut-off. If dose rates exceed the High Radiation Area limits (as defined in Part A of these regulations), emergency shut-off switches shall be located within the high radiation areas so as to be accessible to individuals therein within 10 seconds. These switches and their mode of operation shall be identified by a conspicuously posted sign adjacent to the switch. The emergency shut-off switches shall include a manual reset that must be reset at the switch before x-rays can again be produced from the control panel. After an emergency shut-off switch has been activated, it shall be possible to produce x-rays again only from the control panel.
10.8 Separate Electrical Systems. The interlock system and the emergency shut-off system shall be separate electrical and/or mechanical systems.
10.9 Egress from Shielded Room. A person within the room housing a RGD shall be able to egress at all times.
10.10 Entry into the Shielded Room.
10.10.1 After each exposure and before entry of any personnel, a survey shall be performed upon entry to the shielded room to determine that the RGD is no longer producing radiation.
10.10.2 Personnel devices providing an audible signal when activated by radiation will be acceptable for the survey requirement of H.10i.i.
10.10.2.1 Proper operation of the audible detection device shall be checked daily and a record maintained of this check.
10.10.2.2 The audible device shall be designed so as to clearly indicate entry into a 0.02 mSv (2 mrem) per hour or greater radiation field.
10.10.2.3 All personnel working with the RGD shall be provided with such a device.
10.10.3 Stationary area monitors providing an audible signal when activated by radiation will be acceptable for the survey requirement of Part H, subsection 10.10.1.
10.10.3.1 Proper operation of the stationary detection device shall be checked daily and a record maintained of this check.
10.10.3.2 The stationary device shall be designed so as to clearly indicate entry into a 0.02 mSv (2 mrem) per hour or greater radiation field.
10.10.3.3 Stationary area monitors shall be calibrated annually to determine that the audible signal operates at a 0.02 mSv (2 mrem) per hour radiation field.
10.11 Personnel Monitoring. All personnel associated with the x-ray equipment shall be provided with personnel monitoring devices that shall be calibrated for the x-ray energies being utilized. Records of personnel exposure shall be maintained.
10.12 Training. No registrant shall permit any individual to operate a RGD in a shielded room until such individual has received a copy of, instruction in, and demonstrated an understanding of, operating and emergency procedures for the unit and competence in its use. Records shall be maintained of all operator training.
10.13 Control Panel Security. The equipment control panel shall be provided with a locking device to prevent unauthorized use. Such locking device shall, when locked, prevent the production of radiation by the equipment.
10.14 Malfunctions. If a safety or warning device malfunctions, the control panel shall be locked and tagged "DO NOT USE, Malfunctioning Safety Device" in the "off" position. The control panel shall not be used, except as may be necessary for repair or replacement of the malfunctioning safety or warning device, until the safety or warning device is functioning properly.
11.1 In addition to the General Requirements in Part H, Section 6.0, the following requirements in this section apply. A person requesting Agency approval for a RGD to be used in Personnel Security Screening or Vehicle Screening with intended exposure of human occupants to the primary beam for public protection shall submit in writing the following information to the Agency for evaluation and approval, and show how the dose limits noted below will be met.
11.2 Efficacy Evaluation. An evaluation of all known alternate methods that could achieve the goals of the security screening program, and why these methods will not be used in preference to the proposed approach utilizing ionizing radiation.
11.3 Equipment Evaluation. RGDs used for non-healing arts personnel security screening of humans shall be evaluated annually by a qualified expert for optimization of image quality and radiation dose.
11.4 Dose Limits for General-Use Systems. For general-use screening systems, where system is used without regard to the number of individuals scanned or number of scans per individual in a year, an effective dose for a single complete screening shall be limited to 0.25 uSv (25 urem).
11.5 Dose Limits for Limited-Use Systems. For limited-use screening systems, where equipment is capable of operation greater than 0.25 microsieverts (uSv), or 25 microRem (uRem) per screening, and is used with discretion, the effective dose per screening shall be less than or equal to 0.01 millisieverts (mSv), or 1 milliRem (mRem).
11.6 Dose Limits for Repeat Security Screenings. Individuals subject to repeat security screening at a single venue shall not receive an effective dose greater than 0.25 millisieverts (mSv), or 25 milliRem (mRem) in any one year at the registrant or licensee's facility.
11.7 Vehicle Limitations.
11.7.1 When the procedures for operation of a mobile or fixed RGD used for security screening of vehicles includes knowingly exposing human occupants to the primary beam when screening vehicles, structures or containers, the system shall be subject to the same requirements as general-use or limited-use systems as provided in Part H, subsections 11.2 through 11.6.
11.7.2 If the requirements in Part H, subsection 11.4 through Part H, subsection 11.6 cannot be met if vehicle occupants are knowingly exposed to the primary beam of a security screening system, then there shall be means to assure the occupied portion of the vehicle is outside of the scan area while the primary beam is emitted or procedures shall be established and implemented to assure that no occupants are present in the vehicle during screening.
11.7.3 The effective dose to an individual for a single inadvertent exposure to the primary beam shall not exceed 5 millisieverts (mSv), or 500 milliRem (mRem) and should not exceed 1 millisieverts (mSv), or 100 milliRem (mRem). The reliability of the procedure used to assure that there are no occupants of a vehicle to be scanned shall be commensurate with the potential severity of an inadvertent exposure. If the 5 millisieverts (mSv), or 500 milliRem (mRem) limit cannot be assured, a pre-screening with a mode or system which can meet the limits in Part H, subsection 11.4 through Part H, subsection 11.6 shall be used to verify there are no occupants in the vehicle being examined.
12.1 Any RGD user or manufacturer that cannot meet the applicable requirements of the above sections in this Part shall submit to the Agency a request for an exemption to the specific regulation in question. The exemption request shall demonstrate to the Agency's satisfaction:
12.1.1 That the use of the RGD will not result in undue hazard to public health and safety or property;
12.1.2 That compliance would require replacement or substantial modification of the RGD;
12.1.3 That the registrant will achieve, through other means, radiation protection equivalent to that required by the regulation; and
12.1.4 Why the regulatory standard or requirement could not be met.
NOTICES, INSTRUCTIONS AND REPORTS TO WORKERS; INSPECTIONS
This Part establishes requirements for notices, instructions and reports by licensees or registrants to individuals engaged in activities under a license or registration including but not limited to employees, and options available to such individuals in connection with Agency inspections of licensees or registrants to ascertain compliance with the provisions of the Act and regulations, orders, and licenses issued thereunder regarding radiological working conditions. The regulations in this Part apply to all persons who receive, possess, use, own, sell or transfer sources of radiation registered with or licensed by the Agency pursuant to Parts B and C of these regulations.
General Regulatory Provisions and Specific Requirements
2.1 Each licensee or registrant shall post current copies of the following documents:
2.1.1 The regulations in this Part and in Part D of these regulations;
2.1.2 The license or certificate of registration,
2.1.3 The operating safety procedures applicable to activities under the license or registration; and
2.1.4 Any notice of violation involving radiological working conditions, proposed imposition of administrative penalty, or order issued pursuant to Part A of these regulations, and any response from the licensee or registrant.
2.2 If posting of a document specified in subsections 2.1.1, or 2.1.3 is not practicable, the licensee or registrant may post a notice which describes the document and states where it may be examined.
2.3 Agency Form X "Notice to Workers" shall be posted conspicuously by each licensee or registrant as required by these regulations, see Appendix A.
2.4 Agency documents posted pursuant to subsection 2.1.4 shall be posted conspicuously within 5 working days after receipt of the documents from the Agency; the licensee's or registrant's response, if any, shall be posted within five working days after dispatch from the licensee or registrant. Such documents shall remain posted for a minimum of five working days or until action correcting the violation has been completed, whichever is later.
2.5 Documents, notices, or forms posted pursuant to Section 2.0 shall appear in a sufficient number of places to permit individuals engaged in work under the license or registration to observe them on the way to or from any particular work location to which the document applies, shall be conspicuous, and shall be replaced if defaced or altered.
3.1 All individuals who in the course of employment, education, or training are likely to receive in a year an occupational dose in excess of 1 millisievert (100 mrem):
3.1.1 Shall be kept informed of the storage, transfer, or use of sources of radiation in the licensee's or registrant's workplace;
3.1.2 Shall be instructed in the health protection problems associated with exposure to radiation or radioactive material to the individual and potential offspring, in precautions or procedures to minimize exposure, and in the purposes and functions of protective devices employed;
3.1.3 Shall be instructed in, and instructed to observe, to the extent within the worker's control, the applicable provisions of these regulations and licenses for the protection of personnel from exposures to radiation or radioactive material;
3.1.4 Shall be instructed of their responsibility to report promptly to the licensee or registrant any condition which may constitute, lead to, or cause a violation of the Act, these regulations, or license condition, or any unnecessary exposure to radiation or radioactive material;
3.1.5 Shall be instructed in the appropriate response to warnings made in the event of any unusual occurrence or malfunction that may involve exposure to radiation or radioactive material; and
3.1.6 Shall be advised as to the radiation exposure reports which workers shall be furnished pursuant to Section 4.0.
3.2 In determining those individuals subject to the requirements of subsection 3.1, licensees or registrant must take into consideration assigned activities during normal and abnormal situations involving exposure to radiation and/or radioactive material which can reasonably be expected to occur during the life of a licensed facility. The extent of these instructions shall be commensurate with potential radiological health protection problems present in the workplace.
4.1 Radiation exposure data for an individual and the results of any measurements, analyses, and calculations of radioactive material deposited or retained in the body of an individual shall be reported to the individual as specified in this section. The information reported shall include data and results obtained pursuant to these regulations, orders, or license conditions, as shown in records maintained by the licensee or registrant pursuant to Part D, Section 39.0 of these regulations. Each notification and report shall:
4.1.1 Be in writing;
4.1.2 Include appropriate identifying data such as: the name of the licensee or registrant, the name of the individual, and the individual's unique identification number, such as employee number or Social Security Number;
4.1.3 Include the individual's exposure information; and
4.1.4 Contain the following statement:
"This report is furnished to you under the provisions of the Delaware Radiation Control Regulations, Part J. You should preserve this report for further reference."
4.2 The licensee shall provide an annual report to each individual required to be monitored under these regulations of the dose received in that monitoring year if their occupational dose is in excess of 1 millisievert (100 mrem).
4.3 Each licensee or registrant shall furnish a written report of the worker's exposure to sources of radiation at the request of a worker currently or formerly engaged in activities controlled by the licensee or registrant. The report shall include the dose record for each year the worker was required to be monitored pursuant to Part D, Section 17.0 of these regulations such report shall be furnished within 30 days from the date of the request, or within 30 days after the dose of the individual has been determined by the licensee or registrant, whichever is later. The report shall cover the period of time that the worker's activities involved exposure to sources of radiation and shall include the dates and locations of work under the license or registration in which the worker participated during this period.
4.4 When a licensee or registrant is required pursuant to Sections 46.0, 47.0, 48.0, or 49.0 of these regulations to report to the Agency any exposure of an individual to sources of radiation, the licensee or the registrant shall also provide the individual a written report on the exposure data included therein. Such reports shall be transmitted at a time not later than the transmittal to the Agency.
4.5 At the request of a worker who is terminating employment with the licensee or registrant in work involving exposure to radiation or radioactive material, during the current year, each licensee or registrant shall provide at termination to each such worker, or to the worker's designee, a written report regarding the radiation dose received by that worker from operations of the licensee or registrant during the current year or fraction thereof. If the most recent individual monitoring results are not available at that time, a written estimate of the dose shall be provided together with a clear indication that this is an estimate.
5.1 Each licensee or registrant shall afford to the Agency at all reasonable times opportunity to inspect materials, machines, activities, facilities, premises, and records pursuant to these regulations.
5.2 During an inspection, Agency inspectors may consult privately with workers as specified in Section 6.0. The licensee or registrant may accompany Agency inspectors during other phases of an inspection.
5.3 If, at the time of inspection, an individual has been authorized by the workers to represent them during Agency inspections, the licensee or registrant shall notify the inspectors of such authorization and shall give the workers' representative an opportunity to accompany the inspectors during the inspection of physical working conditions.
5.4 Each worker's representative shall be routinely engaged in work under control of the licensee or registrant and shall have received instructions as specified in Section 3.0.
5.5 Different representatives of licensees or registrants and workers may accompany the inspectors during different phases of an inspection if there is no resulting interference with the conduct of the inspection. However, only one workers' representative at a time may accompany the inspectors.
5.6 With the approval of the licensee or registrant and the workers' representative, an individual who is not routinely engaged in work under control of the licensee or registrant, for example, a consultant to the licensee or registrant or to the workers' representative, shall be afforded the opportunity to accompany Agency inspectors during the inspection of physical working conditions.
5.7 Notwithstanding the other provisions of Section 5.0, Agency inspectors are authorized to refuse to permit accompaniment by any individual who deliberately interferes with a fair and orderly inspection. With regard to areas containing information classified by an Agency of the US Government in the interest of national security, an individual who accompanies an inspector may have access to such information only if authorized to do so. With regard to any area containing proprietary information, the workers' representative for that area shall be an individual previously authorized by the licensee or registrant to enter that area.
6.1 Agency inspectors may consult privately with workers concerning matters of occupational radiation protection and other matters related to applicable provisions of these regulations and licenses to the extent the inspectors deem necessary for the conduct of an effective and thorough inspection.
6.2 During the course of an inspection, any worker may bring privately to the attention of the inspectors, either orally or in writing, any past or present condition which the worker has reason to believe may have contributed to or caused any violation of the Act, these regulations, or license condition, or any unnecessary exposure of an individual to sources of radiation under the licensee's or registrant's control. Any such notice in writing shall comply with the requirements of subsection 7.1.
6.3 The provisions of subsection 6.2 shall not be interpreted as authorization to disregard instructions pursuant to Section 3.0
7.1 Any worker or representative of workers believing that a violation of the Act, these regulations, or license conditions exists or has occurred in work under a license or registration with regard to radiological working conditions in which the worker is engaged may request an inspection by giving notice of the alleged violation to the Office of Radiation Control. Any such notice shall be in writing, shall set forth the specific grounds for the notice, and shall be signed by the worker or representative of the workers. A copy shall be provided to the licensee or registrant by the Office of Radiation Control no later than at the time of inspection except that, upon the request of the worker giving such notice, such worker's name and the name of individuals referred to therein shall not appear in such copy or on any record published, released, or made available by the Agency, except for good cause shown.
7.2 If, upon receipt of such notice, the Office of Radiation Control determines that the complaint meets the requirements set forth in subsection 7.1, and that there are reasonable grounds to believe that the alleged violation exists or has occurred, an inspection shall be made as soon as practicable to determine if such alleged violation exists or has occurred. Inspections pursuant to Section 7.0 need not be limited to matters referred to in the complaint.
7.3 No licensee, registrant, or contractor or subcontractor of a licensee or registrant shall discharge or in any manner retaliate against any worker because such worker has filed any complaint or instituted or caused to be instituted any proceeding under these regulations or has testified or is about to testify in any such proceeding or because of the exercise by such worker on behalf of such worker or others of any option afforded by this Part.
8.1 If the Office of Radiation Control determines, with respect to a complaint under Section 7.0, that an inspection is not warranted because there are no reasonable grounds to believe that a violation exists or has occurred, the Office of Radiation Control shall notify the complainant in writing of such determination. The complainant may obtain review of such determination by submitting a written statement of position with the Division of Public Health. Such Agency will provide the licensee or registrant with a copy of such statement by certified mail, excluding, at the request of the complainant, the name of the complainant. The licensee or registrant may submit an opposing written statement of position with the Division of Public Health. The Division of Public Health will provide the complainant with a copy of such statement by certified mail.
8.2 Upon the request of the complainant, the Division of Public Health may hold a conference in which the complainant and the licensee or registrant may orally present their views. A conference may also be held at the request of the licensee or registrant, but disclosure of the identity of the complainant will be made only following receipt of written authorization from the complainant. After considering all written and oral views presented, the Division of Public Health shall affirm, modify, or reverse the determination of the “Office of Radiation Control” and furnish the complainant and the licensee or registrant a written notification of the decision and the reason therefore.
8.3 All decisions of the Division shall be final and conclusive. Where the licensee or registrant is in disagreement with the action of the Division, the licensee or registrant may appeal the Division’s decision to the Superior Court within 30 days of service or of the postmarked date of the copy of the decision mailed to the licensee or registrant. The appeal shall be on the record to the Superior Court and shall be as provided in §§10142 - 10145 of Title 29.
8.4 If the Office of Radiation Control determines that an inspection is not warranted because the requirements of subsection 7.1 have not been met, the complainant shall be notified in writing of such determination. Such determination shall be without prejudice to the filing of a new complaint meeting the requirements of subsection 7.1.
NOTICE TO WORKERS AGENCY FORM X
STANDARDS FOR PROTECTION AGAINST RADIATION; NOTICES INSTRUCTIONS AND REPORTS TO WORKERS; INSPECTIONS
In Part D of the Delaware Radiation Control Regulations, the Authority on Radiation has established standards for your protection against radiation hazards. In Part J of the Delaware Radiation Control Regulations, the Authority on Radiation Protection has established certain provisions for the options of workers engaged in work under an agency license or registration.
THE REGISTRANT RESPONSIBILITY The Registrant is required to:
1. Apply these regulations to work involving sources of radiation.
2. Post or otherwise make available to you a copy of the Delaware Radiation Control Regulations, and the operating procedures which apply to work you are engaged in, and explain their provisions to you.
3. Post Notice of Violation involving radiological working conditions, and any proposed imposition of administrative penalties and orders.
YOUR RESPONSIBILITY AS A WORKER
You should familiarize yourself with provisions of the Delaware Radiation Control Regulations listed below and facility procedures for safe operation of radiation sources in your workplace. You should observe these provisions for your own protection, and the protection of your co-workers.
WHAT IS COVERED BY THESE REGULATIONS
1. Limits on exposure to radiation and radioactive material in restricted and unrestricted areas;
2. Measures to be taken after accidental exposure;
3. Personnel monitoring, surveys and equipment;
4. Caution signs, labels, and safety interlock equipment;
5. Exposure records and reports;
6. Options for workers regarding Agency inspections; and
7. Related matters.
POSTING REQUIREMENT
COPIES OF THIS NOTICE MUST BE POSTED IN A SUFFICIENT NUMBER OF PLACES IN EVERY ESTABLISHMENT WHERE WORKERS ARE ENGAGED IN ACTIVITIES LICENSED OR REGISTERED, PURSUANT TO PART B OR PART C, BY THE OFFICE OF RADIATION CONTROL, TO PERMIT INDIVIDUALS WORKING IN OR FREQUENTING ANY PORTION OF A RESTRICTED AREA TO OBSERVE A COPY ON THE WAY TO OR FROM THEIR PLACE OF WORK.
REPORTS ON YOUR RADIATION EXPOSURE HISTORY
1. The Delaware Radiation Control Regulations require that the registrant give you a written report if you receive an exposure in excess of any applicable limit set forth in these regulations. The basic limits for exposure to workers are set forth in Part D, Sections 5.0, 6.0, 7.0, 8.0, 9.0 and 12.0 of the regulations.
2. If personnel monitoring is required for your job, and if you request information on your radiation exposures;
(a) The registrant or your employer/or supervisor must advise you annually of your exposure to radiation while you are working, as set forth in Part J, subsections 4.1 and 4.2;
(b) The registrant or your employer/or supervisor must give you a written report, of your radiation exposures upon leaving work in the registered facility as set forth in Part J, subsections 4.3 and 4.5.
INSPECTIONS
All licensed or registered activities are subject to inspection by representatives of the Office of Radiation Control. In addition, any worker or representative of workers who believes that there is a violation of the Delaware Radiation Control Act, the regulations issued thereunder, or the terms of the facility license or registration with regard to radiological working conditions in which the worker is engaged, may request an inspection by sending a notice of the alleged violation to the Office of Radiation Control. The written request must set forth the specific grounds for the notice, and must be signed by the worker as the representative of the workers. During inspections, Agency inspectors may confer privately with workers, and any worker may
bring to the attention of the inspectors any past or present condition which they believe contributed to or caused any violation as described above.
Part J, Agency Form Y Page 1 of 3
INSTRUCTIONS AND ADDITIONAL INFORMATION PERTINENT TO THE COMPLETION OF AGENCY FORM Y
(All doses should be stated in rems)
1. Type or print the full name of the monitored individual in the order of last name
(include "Jr," "Sr," "III," etc.), first name, middle initial (if applicable).
2. Enter the individual's unique identification number, including punctuation. This number should preferably be the employee number (EN). If the EN is unavailable; enter the 9-digit social security number (SSN). If the individual has no social security number, enter the number from another official identification such as a passport or work permit.
3. Enter the code for the type of identification used as shown below:
Code ID Type
EN Employee Number
SSN U.S. Social Security Number
PPN Passport Number
WPN Work Permit Number
OTH Other
4. Check the box that denotes the sex of the individual being monitored.
5. Enter the date of birth of the individual being monitored in the format MM/DD/YYYY.
6. Enter the monitoring period for which this report is filed. The format should be MM/DD/YYYY - MM/DD/YYYY.
7. Enter the name of the licensee or facility not licensed by NRC that provided monitoring.
8. Enter the NRC license number or numbers.
9. Place an "X" in Record, Estimate, or No Record. Choose "Record" if the dose data listed represent a final determination of the dose received to the best of the licensee's knowledge. Choose "Estimate" only if the listed dose data are preliminary and will be superseded by a final determination resulting in a subsequent report. An example of such an instance would be dose data based on self-reading dosimeter results and the licensee intends to assign the record dose on the basis of TLD results that are not yet available.
10. Place an "X" in either Routine or “PSE” (Planned Special Exposure). Choose "Routine" if the data represent the results of monitoring for routine exposures. Choose "PSE" if the listed dose data represents the results of monitoring of planned special exposures received during the monitoring period. If more than one PSE was received in a single year, the licensee should sum them and report the total of all planned special exposures.
Part J, Agency Form Y Page 2 of 3
OF AGENCY FORM Y (Con’t.) All doses should be stated in rems
11. Enter the deep dose equivalent (DDE) to the whole body.
12. Enter the eye dose equivalent (LDE) recorded for the lens of the eye.
13. Enter the shallow dose equivalent recorded for the skin of the whole body (SDE, WB).
14. Enter the shallow dose equivalent recorded for the skin of the extremity receiving the maximum dose
(SDE, ME).
15. Enter the committed effective dose equivalent (CEDE).
16. Enter the committed dose equivalent (CDE) recorded for the maximally exposed organ.
17. Enter the committed dose equivalent (TEDE). The TEDE is the sum of items 11 and 15.
18. Enter the total organ dose equivalent (TODE) for the maximally exposed organ. The TODE is the sum of items 11 and 16.
19. Signature of the monitored individual.The signature of the monitored individual on this form indicates that the information contained on the form is complete and correct to the best of his or her knowledge.
20. [OPTIONAL] Enter the date this form was signed by the monitored individual.
21. Enter the name of the licensee or facility not licensed by the Agency providing monitoring for exposure to radiation (such as a VA facility) or the employer if the individual is not employed by the licensee and the employer chooses to maintain exposure records for its employees.
22. [OPTIONAL] Signature of the person designated to represent the licensee or employer entered in item
23. The licensee or employer who chooses to countersign the form should have on file documentation of all the information on the Agency Form 4 being signed.
24. [OPTIONAL] Enter the date this form was signed by the designated representative.

INSTRUCTIONS AND ADDITIONAL INFORMATION PERTINENT TO THE COMPLETION OF AGENCY FORM Z
(All doses should be stated in rems)
1. Type or print the full name of the monitored individual in the order of last name
(include "Jr," "Sr," "III," etc.), first name, middle initial (if applicable).
2. Enter the individual's unique identification number, including punctuation. This number should preferably be the employee number (EN). If the EN is unavailable the 9-digit social security number (SSN). If the individual has no social security number, enter the number from another official identification such as a passport or work permit.
3. Enter the code for the type of identification used as shown below:
Code ID Type
EN Employee Number
SSN U.S. Social Security Number
PPN Passport Number
WPN Work Permit Number
OTH Other
4. Check the box that denotes the sex of the individual being monitored.
5. Enter the date of birth of the individual being monitored in the format MM/DD/YYYY.
6. Enter the monitoring period for which this report is filed. The format should be MM/DD/YYYY - MM/DD/YYYY.
7. Enter the name of the registered facility not licensed by NRC that provided monitoring.
8. Enter the registration number or numbers.
9. Place an "X" in Record, Estimate, or No Record. Choose "Record" if the dose data listed represent a final determination of the dose received to the best of the licensee's knowledge. Choose "Estimate" only if the listed dose data are preliminary and will be superseded by a final determination resulting in a subsequent report. An example of such an instance would be dose data based on self-reading dosimeter results and the licensee intends to assign the record dose on the basis of TLD results that are not yet available.
10. Place an "X" in either Routine or “PSE” (Planned Special Exposure). Choose "Routine" if the data represent the results of monitoring for routine exposures. Choose "PSE" if the listed dose data represents the results of monitoring of planned special exposures received during the monitoring period. If more than one PSE was received in a single year, the licensee should sum them and report the total of all planned special exposures.
Part J, Agency Form Z
1.1 No person shall use/operate a source of radiation or radiation facility who does not possess a valid certificate, license or registration issued or renewed to that person by the Agency in accordance with Regulation 4481, sections 4.0, 7.0 and 9.0; Part C, sections 1.0 and 33; Regulation 4466, section 7.0 (previously referenced as DRCR B.4, B.7, B.9, C.1, C.33 or RTCR VII A) of the regulations. Only a person who complies with the requirements of the regulations shall be entitled to receive or retain such a certificate, license or registration.
1.2 The owner/manager of a radiation facility shall designate a Radiation Safety Officer (RSO) in accordance with the regulations.
1.3 All facility licenses, registrations and Radiation Technologist/Technician Certificates must be posted in a conspicuous location.
1.4 Any action taken by the Agency against an applicant or certificate, license or registration holder may be appealed to the Authority on Radiation Protection.
“Applicant” means a Person seeking a certificate, license or registration issued under the provisions of the Act and the requirements of the regulations.
“Certificate” is an official document issued by the Agency which authorizes a person to perform a specified radiation activity.
“Exemption” means an exclusion from a regulatory requirement granted by the Agency or Authority. When the exclusion is based on a national standard or similar documented and publicly available information, the Agency may grant it. Otherwise, the exemption shall be referred to the Authority for consideration.
“Hearing” means a proceeding to examine an application or other matter before the Authority in order to adjudicate rights, duties, or privileges.
“Imminent Radiation Hazard(s)” means an imminent hazard exists when the radiation levels that exist are in excess of three times the regulatory limit.
“License” means a license issued by the Agency in accordance with the regulations.
“Licensee” means any person who is licensed by the Agency in accordance with the regulations and the Act.
"Licensed Practitioner" means an individual licensed to practice medicine, dentistry, dental hygiene, podiatry, chiropractic, advanced practice nursing, or osteopathy in this State.
“Licensee's Representative” means a person who has been authorized by the licensee to represent them during activities or proceedings governed by the regulations.
“Modification” means a change in the specification of a machine or radiation facility.
“Notice of violation” means a written statement of one or more alleged infringements of a legally binding requirement. The notice normally requires the licensee, registrant, other permit holder to provide a written statement describing the following:
corrective steps taken by the licensee, registrant, or other permit holder, and the results achieved;
corrective steps to be taken to prevent recurrence; and
the projected date for achieving full compliance. The Authority may require responses to notices of violation to be under oath.
1. Has earned a master's and/or doctoral degree in physics, medical physics, biophysics, radiological physics, medical health physics, or equivalent disciplines from an accredited college or university; and
2. Has been granted certification in the specific subfield(s) of medical physics with its associated medical health physics aspects by an appropriate national certifying body and abides by the certifying body's requirements for continuing education; and/or
3. Is credentialed in accordance with Part X, subsection 3.4, as amended.
“Registration” means to enroll or register with the Agency in accordance with the regulations.
“Regulations” mean all parts of the Delaware Radiation Control Regulations (DRCR) and all parts of the Delaware Radiation Technologist Certification Regulations (RTCR).
“Severity level” means a classification of violations based on relative seriousness of each violation and the significance of the effect of the violation on the occupational or public health or safety.
“Violation” means an infringement of any rule, certificate, license or registration condition, order of the agency, or any provision of the Act.
3.1 Certificate
3.1.1 A Radiation Technologist Certificate is required to practice radiation technology (Request Application Form ORC-R16) as outlined in the Delaware Radiation Technologist Certification Regulations, Regulation 4466.
3.1.2 A Plan Approval Certificate is required for the construction or modification of a radiation facility (Request Shielding Information Letter) as outlined in the regulations.
3.2 License
3.2.1 A Radioactive Material License is required to use and/or possess a radioactive material source (Request Application Form ORC-R2).
3.3 Registration
3.3.1 A Registration is required to possess and/or use a radiation machine (i.e. x-ray equipment) or operate a radiation machine facility. (Request Application Form ORC-R1)
3.3.2 Registration is required to possess a source of Radioactive Material. (Request Application Form ORC-R2)
3.3.3 A Registration is required to perform a radiation service including but not limited to, repair, install or calibrate radiation equipment/devices, perform health physics or radiation protection consultations or surveys, personnel dosimetry and therapeutic radiation physics (Request Application Form ORC-R3).
3.4 Annual/Biennial License or Registration
3.4.1 An annual or biennial license or registration shall be issued to any person desiring to possess, use, provide or operate a source of radiation, radiation facility or radiation service in the State for more than thirty (30) days upon written application to the Agency.
3.4.2 The Agency shall issue a license or registration to the applicant if the Agency's inspection or examination reveals that the proposed facility, use, source of radiation and/or individual complies with the requirements of the regulations.
3.4.3 An annual/biennial license or registration is valid for one (1) or two (2) anniversary year(s) from the date of issuance, unless a new owner, management, firm or lessee takes possession of the facility, source of radiation or service; or the license or registration is revoked by the Authority for violations of the regulations. A license or registration is not transferable.
3.5 Temporary Use of Out of State Source
3.5.1 This license or registration allows for the temporary use of an out-of-state radiation source in Delaware.
3.5.2 X-ray equipment - send advance written notice to the Agency as per Regulation 4481 (Part B).
3.5.3 Radioactive Material Source/Device - send advance written notice to the Agency of the intended use and location with photo copy of NRC or Agreement State License as per Part C.
4.1 A valid certificate, license or registration is not transferable. Therefore, it is the responsibility of the new owner/manager to acquire an operating certificate, license or registration prior to commencing operations.
4.2 New construction or modification of an existing room or area associated with a source of radiation requires plan approval in accordance with Regulation 4481.
4.3 The owner/manager of a radiation source is responsible for notifying the Agency prior to its sale, transfer, lease, or disposal in accordance with requirements listed in Regulations 4481 (Part B).
4.4 If any renovations or modifications of the physical structure of the existing facility are required, based on current or previous inspection reports of the Agency, the new owner/manager will be held responsible for these renovations or modifications.
4.4.1 Completion of any renovations shall be achieved prior to the start of operation, unless the new owner/manager is granted an exemption in accordance with Regulation 4480.
5.1 Inspection Frequency.
5.1.1 An inspection of a registered facility shall be performed at least every two (2) years for medical facilities utilizing angiography, radiography, fluoroscopy, computed tomography (CT), mammography, stereotactic breast biopsy systems and radiation therapy modalities, and at least every four (4) years for other registered facilities, including dental, bone densitometry, podiatry, veterinary, academic and industrial.
5.1.2 Additional inspections of registered facilities as described in the regulations shall be performed as often as necessary to ensure and verify compliance with the regulations.
5.2 Access
5.2.1 The Authority or its duly authorized representatives shall have the power to enter at all reasonable times upon any private or public property for the purpose of determining whether or not there is compliance with or violations of 16 Del.C., Ch.74 and rules and regulations issued thereunder, except that entry into areas under the jurisdiction of the federal government shall be effected only with the concurrence of the federal government or its duly authorized designated representative.
5.2.2 The Agency may suspend for a period not to exceed (30) days, the certificate, license or registration to operate/use a source of radiation for refusing access to the representative(s) of the Agency if the Agency can show good cause that there is a risk of imminent harm to the public from ionizing radiation at the facility to which the Agency is attempting access.
5.3 Inspection Report Form
5.3.1 Facility Inspection Form ORC-R11 shall be used to record the results of inspections at registered radiation source facilities as specified in (Part K), Compliance Procedures.
5.3.1.1 The co-signed original of the completed inspection report form shall be furnished to the person named on the license or registration or the Radiation Safety Officer (RSO) at the conclusion of the inspection.
5.3.1.2 The completed inspection form shall list the violation(s) (if any), give the time period for correcting the violation(s) and state the corrections to be made. The inspection report form shall summarize the requirement(s) of the regulations.
5.3.1.3 The form shall also state that "Failure to comply with time limits for correction of any violations cited in this notice shall result in automatic license or registration suspension and immediate cessation of use of a source radiation or radiation area in accordance with Compliance Procedures of the regulations of the Authority on Radiation Protection."
5.3.2 The completed inspection report form is a public document that shall be made available for public disclosure to any person who requests it in accordance with the "Freedom of Information Act 29 Del.C., Ch. 100."
5.3.3 A Notice of Violation (Form ORC-R10) must be posted in a conspicuous location until the violation has been corrected.
5.4 Types of Inspections. Inspections are performed to verify compliance with all applicable laws and regulations.
5.4.1 Regular Inspections
5.4.1.1 Regular inspections are performed on a routine basis in permanent, operating, registered facilities. These inspections shall address all items on the inspection report form. Items in violation shall be recorded by item number.
5.4.2 Follow-up Inspections
5.4.2.1 Follow-up inspections shall be performed when a regular inspection finds one (1) or more Severity Level 1 violation(s) or three (3) or more Severity Level 2 violations.
5.4.2.2 Follow-up inspections may also be performed to verify proper posting of certificates, licenses or registrations, after complaint and investigation inspections, or after administrative hearings.
5.4.3 Complaint Inspections
5.4.3.1 Complaint inspections are performed in response to formal or informal complaints against registered facilities.
5.4.3.2 A complete inspection may be performed by the Agency in the interest of protecting the public.
5.4.4 Investigation Inspections
5.4.4.1 Investigation inspections are performed on non-registered radiation facilities for determining whether compliance with the regulations is required.
5.4.5 Other Inspections
5.4.5.1 Other inspections include construction/modification, pre-operational and other inspections not included above.
6.1 Violations of the regulations have been classified as Severity Level 1 and Severity Level 2 depending upon the impact of the violation.
6.1.1 Severity Level 1 violations generally could result in overexposure to the patient or operator or violate individual's rights as outlined in the regulations.
6.1.2 Severity Level 2 violations generally will not result in overexposure but may indicate a lack of administrative controls over the use of the radiation source. Reference Appendix A for violation classifications.
6.2 Severity Level 1 Items
6.2.1 When one (1) or two (2) Severity Level 1 items of violation are found by any inspection, the related source(s) of radiation shall be tagged "out-of-use." All violations shall be corrected prior to returning the unit in service.
6.2.2 When three (3) or more Severity Level 1 items of violation are found by any inspection, the certificate, license or registration shall be suspended in accordance with Compliance Procedures. All violations shall be corrected prior to resuming registered activities.
6.2.3 The licensee or registrant shall inform the Agency in writing within 10 days of issuance of the inspection report of the proposed method or means of correcting the Severity Level 1 violation(s) and of the date when the correction will be made.
6.2.4 Follow-up inspections shall be conducted within 30 days to assure correction.
6.3 Severity Level 2 Items
6.3.1 All Severity Level 2 items shall be corrected as soon as possible, but in any event, within 60 days.
6.3.2 Follow-up inspections shall be conducted within 60 days to assure corrections have been completed.
6.4 If a follow-up inspection of a registered facility indicates non-compliance of a previously cited violation of the last inspection, a hearing before the Authority on Radiation Protection shall be scheduled. Additionally, the Agency may file a complaint to the Authority.
7.1 Radiation Machine Facility Permit Fees are established for issuance of annual registration permits to radiation machine facilities located within the State of Delaware, in accordance with 16 Del.C., Ch 74.
7.2 Fee Schedule
Category I: Facilities with a total of five or more of the medical modalities or non-medical modalities listed below: $1370.
Category II: Facilities with a total of three or four of the medical modalities or non-medical modalities listed below: $1030.
Category III: Facilities with two of the medical modalities listed below: $690.
Category IV: Facilities with one of the medical modalities listed below, and an annual patient workload of 750 examinations or more: $275.
Category V: Facilities with one of the medical modalities listed below, and an annual patient workload of less than 750 examinations, and all other radiation installations with one or two of the non-medical modalities listed below except as listed under Category VI: $140.
Category VI: Dental, podiatric, bone densitometry or veterinary installations: $75.
7.3 Fee Category Definitions
7.3.1 For purposes of the fee schedule set out in 7.2 above, the following definitions shall apply:
“medical modalities” shall mean radiography, fluoroscopy, computed tomography, angiography, stereotactic breast biopsy systems, and radiation therapy, utilized on humans.
7.3.2 For purposes of the fee schedule set out in 7.2 above, the following definitions shall apply: “non-medical modalities” shall mean radiography, fluoroscopy, analytical equipment (including electron microscopes, fluorescence analysis and x-ray diffraction equipment), computed tomography, and particle accelerators, not utilized on humans.
8.1 If the Agency determines that condition(s) exist(s) in a registered facility which represent(s) a threat to life or a serious risk of damage to health, safety and welfare of the workers or public, or if serious violations, repeat violations, or general disregard of accepted radiation practice are found to exist, administrative action is required.
8.2 Compliance Conference. A meeting held by the Agency with management of a licensee, registrant, or other license, certificate or registration holder to discuss the following:
8.2.1 safety, safeguards, or environmental problems;
8.2.2 compliance with regulatory, license condition, or registration condition requirements;
8.2.3 proposed corrective measures including, but not limited to, schedules for implementation; and
8.2.4 enforcement options available to the agency.
8.3 Suspension of Certificate, License or Registration
8.3.1 Conditions for Suspension of Certificate, License or Registration
8.3.1.1 If some condition(s) is/are determined to exist in a registered facility which present(s) an imminent radiation hazard to human health, the Agency may cease operations of the source of radiation without a hearing or written notice until such time the conditions have been corrected.
8.3.1.2 Further enforcement action shall be taken in accordance with the regulations.
8.3.1.3 Such an imminent radiation hazard shall include, but is not limited to, any one of the following:
8.3.2 The suspension shall be effective upon receipt of written notice by the Radiation Safety Officer or the person in charge of the radiation facility or their agent.
8.3.2.1 A suspension statement recorded on the inspection report by the Agency constitutes a written notice.
8.3.2.2 Service of a written notice of suspension by the Agency stating the reason(s) for the suspension must be made by the close of the following business day.
8.3.2.3 The certificate, license or registration shall not be suspended for a period longer than necessary to correct the hazardous conditions, unless mutually agreed upon.
8.4 Right of Appeal of Suspension
8.4.1 The owner/manager of the registered facility may submit in writing an appeal to the Authority on Radiation Protection for reconsideration of a decision by the Agency.
8.4.2 The notice of appeal shall be sent via certified mail to the Authority on Radiation Protection and to the Agency. An appeal shall not automatically stay the decision of the Agency.
8.4.3 After review for potential radiation hazard to the public, the suspended or revoked license or registration may be stayed on the order of the Program Administrator for Radiation Control.
8.4.4 If a notice of appeal is not filed within thirty (30) days, the license or registration suspension or revocation recommendation shall be upheld and other enforcement action taken in accordance with the this regulation, section 9.4. If the notice of appeal is timely filed, the Authority on Radiation Protection shall hold a hearing at its earliest opportunity.
8.5 Reinstatement of Certificate, License or Registration:
8.5.1 In consultation with the Authority on Radiation Protection, if a follow-up inspection by a representative of the Agency shows the imminent radiation hazard(s) to human health no longer exist(s), the suspension shall be lifted immediately and the certificate, license or registration returned.
8.5.2 If there is no evidence that the imminent radiation hazard(s) has/have been corrected, the suspension will remain in effect until the condition(s) has been corrected.
8.5.3 The owner/manager of the registered facility may request, in writing, a hearing before the Authority on Radiation Protection at any time during the period of suspension, for the purpose of demonstrating that the imminent radiation hazard(s) no longer exist.
8.5.4 The request for hearing shall not stay the suspension.
8.5.5 A record of all proceedings shall be made in accordance with Compliance Procedures.
8.6 Exemption Requests
8.6.1 All requests for exemptions must be filed with the Agency for review. If a determination cannot be made by the Agency, the exemption request must be referred to the Authority.
8.6.2 Agency may grant, an exemption if based on national standards.
8.6.3 Shall hear an appeal by the applicant within ten (10) days following the denial of an exemption request by the Agency. If an exemption is granted; record of the action shall become a part of the permanent record of the facility.
8.6.4 An exemption is not transferable.
9.1 Administrative Hearings
9.1.1 The Authority may, upon sworn complaint or upon its own initiative, cause an investigation to be held to determine whether a license or registration holder, former license or registration holder or applicant has engaged in any activity requiring disciplinary action.
9.1.2 Upon completion of said investigation, the Authority shall hold a hearing to determine whether a license or registration holder, former license or registration holder or applicant has engaged in activities specified in this section as grounds for disciplinary action.
9.1.3 The Authority shall fix the time and place for the hearing.
9.2 The Authority shall cause a copy of the charges, together with a notice of the time and place for the hearing, to be served on the alleged violator at least 30 days prior to the date fixed for the hearing.
9.2.1 When personal service cannot be effected, the Agency shall mail a copy of the charges and of such notice to the alleged violator at his last known address according to the records of the Agency.
9.3 In all proceedings herein:
9.3.1 The alleged violator may be represented by counsel who shall have the right of examination and cross-examination.
9.3.2 The alleged violator and the Agency may call witnesses and admit documentary evidence on their own behalf.
9.3.3 Testimony before the Authority shall be under oath. Any member of the Authority shall have power to administer oaths for this purpose.
9.3.4 A record of the hearing shall be made.
9.3.4.1 At the request and expense of either party such record shall be transcribed with a copy to the other party.
9.3.5 The decision of the Authority shall be based upon a preponderance of the evidence.
9.3.5.1 If the charges are supported by such evidence, the Authority may revoke, refuse to issue, or suspend a certificate, license or registration, or otherwise discipline the individual.
9.3.5.2 A suspended certificate, license or registration may be reissued upon a further hearing initiated at the request of the suspended licensee by written application in accordance with the rules of the Authority and if the Authority finds compliance has been achieved.
9.4 Revocation and Appeal of Suspended Certificate, License or Registration
9.4.1 The Authority on Radiation Protection, at its earliest opportunity, shall consider the Agency's recommendation for certificate, license or registration revocation or hear an appeal by the owner/manager whose permit stands suspended.
9.4.2 The Authority on Radiation Protection shall, at each scheduled meeting, release the name(s) and address(es) of those registered facilities currently meeting the following criteria:
9.4.3 Certificate, License or Registration permanently revoked
9.4.4 Certificate, License or Registration suspended
9.4.5 Certificate, License or Registration censored
9.4.6 Issued a letter of reprimand
9.4.7 Certificate, License or Registration application refused
9.4.8 Certificate, License or Registration renewal refused
9.4.9 Use of source of radiation terminated
9.5 Exemptions referred to the Authority
9.5.1 The Authority on Radiation Protection:
9.5.2 May from time to time grant written permission to vary from particular provisions set forth in the regulations when the extent of the variation is clearly specified and it is documented to the Authority's satisfaction that:
9.5.2.1 Such variation is necessary to obtain a beneficial use by the owner/manager of an existing facility;
9.5.2.2 Appropriate alternative measures have been taken to protect the health and safety of the public from ionizing radiation and assure that the purpose of the provisions from which the variation is sought will be observed.
9.5.3 The Authority shall hear an appeal by the applicant within thirty (30) days following the denial of an exemption request by the Agency. If an exemption is granted; record of the action shall become a part of the permanent record of the facility.
9.5.4 An exemption is not transferable.
10.1 Inspection/Enforcement
10.1.1 A registered facility may be inspected by the Agency as often as necessary for enforcement of the regulations.
10.1.2 Registered facilities, their employees and their agents shall be in compliance with the regulations.
10.1.3 The established administrative procedures for the implementation and enforcement of the provisions and penalties of 16 Del.C. Ch, 74, shall be applicable to this section.
10.2 Failure to allow access, inspection or tests by the Agency representative(s) shall cause the Agency to prohibit the use of a source of radiation, close the registered facility, and/or suspend the facility certificate, license or registration if the Agency can show good cause to believe that there is a risk of immediate harm to the public from ionizing radiation at the facility to which the Agency is attempting access.
10.3 Procedure when Overexposure/Radiation Contamination is Suspected:
10.3.1 When the Agency has reasonable cause to suspect possible individual overexposure or radioactive contamination at a registered facility in excess of limits set forth in regulation 4483 (Part D) it may conduct a radiation investigation which can indicate exposure histories of individuals or make any other investigations as indicated and shall take appropriate action.
10.3.2 The Agency may require any or all of the following measures:
10.3.2.1 The immediate closing of the registered facility or prohibition of the use of a radiation source or radiation area until, in the opinion of the Agency, no further danger of overexposure or contamination exists.
10.3.2.2 Restriction of an employee(s) services to some area of the radiation facility where there would be no opportunity to use a source of radiation or be irradiated.
10.3.2.3 Any other action which the Agency can demonstrate is necessary to protect the health of the public and other employees of the radiation facility.
11.1 Any person who violates a provision of the regulations and any person who is the holder of a certificate, license or registration or who otherwise operates a registered facility that does not comply with the requirements of the regulations shall be subject to the provisions of 16 Del.C. section 7416.
11.2 Operation without a Certificate, License or Registration:
11.2.1 If a facility or individual is found operating without a valid certificate, license or registration as required by Regulation 4481 (Part B) or Regulation 4466, section 7.0 (previously referenced as DRCR B.5. or RTCR, section 7.0 of the regulations), the Agency may act on behalf of the Authority and the source of radiation shall be tagged out-of-use.
11.3 The Agency may seek to enjoin violations of the regulations.
11.4 A conspicuous notice shall be prominently displayed on the radiation source or at all entrances of facilities meeting the following criteria:
11.4.1 Failed to obtain a valid certificate, license or registration; or
11.4.2 Certificate, License or Registration suspended; or
11.4.3 Certificate, License or Registration revoked.
Violation Classification (Typical/Not all inclusive)
Radiation Source Facility
Severity Level 1 | Severity Level 2 | ||
1. | Operating without a permit, (PART K), section 1.1 | 1. | Registration Form 16. Del.C. 7402, (PART K), section 1.0 |
2. | Personnel Overexposure, Regulation 4483, section 6.0 (D. 201) | 2. | Notice to Employees, Regulation 4489, section 2.3 (J.11) |
3. | Radiation Levels Excessive, Regulation 4483, sections 20.0 and 21.0 (D.601,D.602,D.603) | 3. | Safety Procedures, (PART F), section 3.1 (D.101,D.1102) |
4. | Personnel Monitoring, Regulation 4483, section 18.0 (D.502) | 4. | Personnel Monitoring Records, Regulation 4483, section 46.0 (D.1107) |
Medical Uses
Severity Level 1 | Severity Level 2 | ||
1. | Patient exposure (ESE) not in accordance with (PART F), section 3.1.4 (F.3.a.iv) | 12. | |
2. | Tube support not in accordance with (PART F), section 4.7 (F.4.g) | 13. | Maintenance of x-ray system records not in accordance with (PART F), section 3.1.7 (F.3.xiv) |
3. | Technique indicators not in accordance with (PART F), section 4.8 (F.4.h) | 14. | Speed of film/screen not in accordance with (PART F), section 3.1.9 (F.3.ix.1) |
4. | Multiple tube indication not in accordance with (PART F), section 4.6 (F.4.f) | 15. | Technique chart not in accordance with (PART F), section 3.1.3 (F.3.a.iii) |
5. | Gonadal shielding not in accordance with (PART F), section 3.1.6 (F.3.a.vi) | 16. | Warning label not in accordance with (PART F), section 4.1 (F.4.a) |
6. | Operator apron/barrier not in accordance with (PART F), section 3.1.5.2 (F.3.viii.6) | 17. | Patient dose of 1 gray (100 rads) or more not reported to the Agency, (PART F), section 5.11 (F.5.k) |
Radiographic
Severity Level 1 | Severity Level 2 | ||
1. | Location of x-ray controls not in accordance with (PART F), sections 6.25; 7.35 (F.6.b.v and F.7.c.v.1) | 1. | Visual/audio signal not in accordance with (PART F), sections 6.22; 7.32 (F.6.b.ii and F.7.c.ii) |
2. | Position indicating device not in accordance with Regulation 4480, section 6.3 (F.4.j) | 2. | Adjustment of x-ray field not in accordance with (PART F), section 6.11 (F.5.iii) |
3. | Beam Collimation not in accordance with (PART F), section 7.2 (F.6.h.i) | 3. | Indication of field size, upon adjustments, not in accordance with (PART F), section 6.1.2 (F.6.a.ii2) |
4. | Filtration deficiency of 0.2+ mm not in accordance with (PART F), section 4.5.1 (F.4.e.ii) | 4. | Means to limit source to skin distance not in accordance with (PART F), section 7.1 (F.7.a) |
5. | Variation in timer linearity of 15% or more not in accordance with (PART F), sections 6.2.4; 7.3.4 (F.6.b.iv and F.6.g) | 5. | Filtration deficiency of 0.2 mm or less not in accordance with (PART F), section 4.5.1 (F.4.e.ii) |
6. | Variation in exposure reproducibility of 15% or more not in accordance with (PART F), sections 6.4; 7.4 (F.6.d and F.7.d) | 6. | Variation in timer reproducibility linearity more that 10% but less than 15% not in accordance with (PART F), section 6.2.4 (F.6.b.iv) |
7. | Total misalignment of x-ray/light field edges of 5% or more not in accordance with (PART F), section 6.1.2.1 (F.6.i.2) | 7. | Variation in exposure reproducibility more than 5% but less than 10% not in accordance with (PART F), sections 6.4; 7.4 (F.7.d and F.6.vii.d) |
8. | Total misalignment of x-ray beam/image receptor centers of 5% or more not in accordance with (PART F), section 6.1.2.1 (F.6.i.2) | 8. | Total misalignment of light/x-ray field edges more than 2% but less than 5% not in accordance with (PART F), section 6.1.1.2 (F.6.a.iv.i) |
9. | Discorrespondence of indicated x-ray field with beam limited x-ray field of 5% or more not in accordance with (PART F), section 6.1.2.3. (F.6.a) | 9. | Total misalignment of x-ray beam/image receptor centers more than 2% but less than 5% not in accordance with (PART F), section 6.1.2.1 (F.6.a.iv.2) |
10. | Discorrespondence of indicated x-ray with beam limited x-ray field more than 2% but less than 5% not in accordance with (PART F), section 6.2.3 (F.6.a.i.2) | ||
Fluoroscopic
Severity Level 1 | Severity Level 2 | ||
1. | Activation of x-ray production not in accordance with (PART F), section 5.4. (F.5.b) | 1. | Posting of exposure rate measurements not in accordance with (PART F), section 5.3 (F.5) |
2. | Annual exposure rate measurement not in accordance with (PART F), section 5.3.1.4 (F.5.4) | 2. | Measurement records and posting of same not in accordance with (PART F), section 5.3 (F.5) |
3. | Exposure rate not in accordance with (PART F), section 5.3 (F.5.c) | 3. | Adjustment of field not in accordance with (PART F), section 5.1.2 (F.5.a.ii) |
4. | Useful beam protective barrier not in accordance with (PART F), section 5.4 (F.5.d) | 4. | Means of field adjustment not in accordance with (PART F), section 5.1.2 (F.5.a.ii) |
5. | Limitation of x-ray field to image receptor not in accordance with (PART F), section 5.1 (F.5) | 5. | Indication of kV and mA not in accordance with (PART F), section 5.5 (F.5.4) |
6. | Minimum field at maximum SID not in accordance with (PART F), section 5.1.2.2.2 (F.5.iii3) | 6. | Means to indicate that beam is perpendicular to image receptor not in accordance with (PART F), section 5.1 (F.5.2.d) |
7. | High level control exposure rate limit not in accordance with (PART F), section 5.3 (F.5.c) | 7. | Audible signal to reset not in accordance with (PART F), section 5.7 (F.5.c.b and F.5.g) |
8. | Timing device not in accordance with (PART F), section 5.7 (F.5.g) | 8. | Minimum source to skin distance not in accordance with (PART F), section 5.6 (F.5.f) |
9. | Control of scattered radiation not in accordance with (PART F), section 5.8 (F.5.h) | 9. | Exposure rate due to transmission through primary barrier not in accordance with (PART F), section 5.4 (F.5.a.1) |
10. | Failure to report patient dose of 1+ gray to the Agency, (PART F), section 5.11 (F.5.k) | ||
Industrial Radiographic
Severity Level 1 | Severity Level 2 | ||
1. > 2 mR/hr. | Failure to maintain radiation level from device < 4" in diameter below maximum allowable limit of 50 mR/hr. at 6" in accordance with Regulation 4484, section 5.1 (E.101) | < 2 mR/hr. | |
2. > 2 mR/hr. | Failure to maintain radiation level from device > 4" diameter below maximum allowable limit of 200 mR/hr. at surface in accordance with Regulation 4484, section 5.1 (E.101) | < 2 mR/hr. | |
3. > 2 mR/hr. | Failure to maintain radiation level from storage container below maximum allowable limit of 200 mR/hr. at the surface in accordance with Regulation 4484, section 5.1 (E.101) | < 2 mR/hr. | |
4. > 2 mR/hr. | Failure to maintain radiation level from storage container below maximum allowable limit of 10 mR/hr. at a distance of 1 meter in accordance with Regulation 4484, section 5.1 (E.101) | < 2 mR/hr. | |
Locking of Source | |||
5. | Radiation Source Storage Container was found unlocked and not under the direct surveillance of authorized individual in accordance with Regulation 4484, section 5.2.1 (E.102) | ||
6. | Exposure device and/or storage container was not locked during transit or prior to unit being made secure as required by Regulation 4484, section 5.2.2 (E.103) | ||
7. | Device was not physically secured to prevent tampering or removal as required by Regulation 4484, section 5.3 (E.103) | ||
Survey Instruments
Severity Level 1 | Severity Level 2 | ||
1. | Failure to adequately maintain survey instruments in accordance with Regulation 4484, section 5.4.1 (E.104) | 1. | Failure to possess calibrated back up instrument Regulation 4484, section 5.4.1 (E.104) |
2. | Survey meter not capable of detecting 2-1000 mR/hr. as required by Regulation 4484, section 5.4.1 (E.104) | 2. | Failure to conduct quarterly calibrations in accordance with Regulation 4484, section 5.4.2 (E.106) |
3. | Failure to calibrate unit after servicing in accordance with Regulation 4484, section 5.4.2 (1st bullet point) (E.104) | ||
4. | Failure to achieve accuracy of (+ 20%) as required by Regulation 4484, section 5.4.2 (2nd bullet point) (E.104) | ||
5. | Failure to calibrate each scale using 2 points other than zero as required by Regulation 4484, section 5.4.2 (3rd bullet point) (E.104) | ||
6. | Failure to maintain calibration records for two years in accordance with Regulation 4484, section 5.4.3 (E.107) | ||
Leak Testing/Repair
Severity Level 1 | Severity Level 2 | ||
1. | Service to sealed source not conducted by authorized individual in accordance with Regulation 4484, section 5.5.1 (E.105) | 1. | Failure to conduct leak test within 6 month interval as required by Regulation 4484, section 5.5.2 (E.105) |
2. | Failure to immediately withdraw contaminated unit from service to effect decontamination/repair as required by Regulation 4484, section 5.5.4 (E.105) | 2. | Leak test procedure not capable of detecting unit 0.005 uCi of removable contamination as required by Regulation 4484, section 5.5.2 (E.105) |
3. | Failure to properly label unsecured source in accordance with Regulation 4484, section 5.5.5 (E.105) | 3. | Failure to record test results in microcuries as required by Regulation 4484, section 5.5.2 (E.105) |
4. | Failure to notify agency within 5 days of receiving test results as required by Regulation 4484, section 5.5.4 (E.105) | ||
Inventory/Inspection
Severity Level 1 | Severity Level 2 | ||
1. | Failure to perform quarterly operational safety maintenance and inspection in accordance with Regulation 4484, section 5.8.2 (E.108) | 1. | Failure to conduct quarterly inventory as required by Regulation 4484, section 5.6 (E.106) |
2. | Failure to withdraw defective unit from service as required by Regulation 4484, section 5.8.3 (E.109) | 2. | Failure to maintain utilization log as required by Regulation 4484, section 5.7 (E.107) |
3. | Failure to conduct and record alarm tests at the beginning of each period of use as required by Regulation 4484, section 5.9.2 (E.109, E.202) | 3. | Failure to log required information in accordance with Regulation 4484, sections 5.7.1 - 5.7.4 (E.107) |
4. | Failure to properly document quarterly inspection and maintenance of radiation devices in accordance with Regulation 4484, section 5.8 (E.108) | ||
5. | Failure to record and maintain test results in accordance with Regulation 4484, section 5.9 (E.303) | ||
Limitations/Procedures/Controls
Severity Level 1 | Severity Level 2 | ||
1. | Individual is not qualified Industrial Radiographer as defined by Regulation 4484, sections 10.1.1.1 - 10.1.1.3 (E.201) | 1. | Failure to maintain training and test records in accordance with Regulation 4484, section 10.1.3 (E.201) |
2. | Individual is not a qualified radiographer's assistant trainee as defined by Regulation 4484, section 10.1.2 (E.201) | 2. | Failure to record dosimetry data in accordance with Regulation 4484, section 10.3.2 (E.201) |
3. | Qualified operator(s) not wearing personal monitoring devices as required by Regulation 4484, section 10.3.1 (E.203) | ||
4. | Operating and Emergency procedures do not meet the requirements of Regulation 4484, sections 10.2.1.1 - 10.2.1.7 (E.203) | ||
Precautionary Procedures
Severity Level 1 | Severity Level 2 | ||
1. | Failure of qualified operators to maintain direct surveillance in accordance with Regulation 4484, section 11.1 (E.301) | 1. | Failure to post required information in accordance with Regulation 4484, section 11.2 (E.302) |
2. | Failure to have calibrated and operable survey instrument available in accordance with Regulation 4484, section 11.3.1 (E.303) | 2. | Failure to maintain survey records for 2 years in accordance with Regulation 4484, section 11.3.6 (E.303) |
3. | Failure to conduct physical radiation survey in accordance with Regulation 4484, section 11.3.2 and/or 11.3.3 (E.303) | ||
4. | Failure to maintain records at temporary job site in accordance with Regulation 4484, sections 11.4.1 - 11.4.6 (E.304) | ||
Special Requirements
Severity Level 1 | Severity Level 2 | ||
1. | Failure to meet requirements for enclosed radiography in accordance with Regulation 4484, section 11.5.1 (E.306) | 1. | Failure to maintain evaluation records for 2 years in accordance with Regulation 4484, section 9.1.2 (E.306) |
2. | Failure to meet exemption requirements and maintain unit in accordance with Regulation 4484, section 11.5.2 (E.306) | ||
Analytical
Severity Level 1 | Severity Level 2 | ||
1. | Failure to provide safety device in accordance with Regulation 4487, section 3.1 (H.3) | 1. | Failure to label equipment in accordance with Regulation 4487, section 3.4 (H.3) |
2. | Failure to provide warning devices in accordance with Regulation 4487, section 3.2 (H.3) | 2. | Failure to have appropriate warning lights in accordance with Regulation 4487, section 3.2 (H.3) |
3. | Failure to install shutter in accordance with Regulation 4487, section 3.5 (H.3) | 3. | Failure to provide protective cabinet to prevent leakage in accordance with Regulation 4487, section 3.7 (H.3) |
4. | Failure to construct tube housing in manner which ensures compliance with Regulation 4487, section 3.6 (H.3) | 4. | Failure to conduct Radiation Surveys in accordance with Regulation 4487, section 4.2 (H.4) |
5. | Failure to obtain Radiation Safety Officer's approval for by-passing safety device in accordance with Regulation 4487, sections 5.1 and 5.2 (H.5) | 5. | Failure to document survey record in accordance with Regulation 4487, section 4.2 (H.4) |
6. | Failure to provide and/or ensure the use of extremity dosimeters in accordance with Regulation 4487, section 6.2 (H.6) | 6. | Failure to post area in accordance with Regulation 4487, section 4.3 (H.4) |
7. | Failure to instruct and/or ensure operator competence in accordance with Regulation 4487, section 6.1 (H.6) | 7. | Failure to secure unused ports in accordance with Regulation 4487, section 3.3 (H.3) |
Particle Accelerators
Limitations
Severity Level 1 | Severity Level 2 | ||
1. | Operator did not receive radiation safety instruction or could not demonstrate understanding of radiation safety Regulation 4488, section 6.1 (I.6) | 1. | Operator had not received copies Regulations 4483, 4488, 4489 & emergency procedures, Regulation 4488, section 6.1 (I.6) |
2. | Operator unable to demonstrate competency in the use of the accelerator Regulation 4488, section 6.1 (I.6) | 2. | Radiation Safety Committee/Radiation Safety Officer is not authorized to terminate operations as per Regulation 4488, section 6.2 (I.6) |
Controls/Interlocks
Severity Level 1 | Severity Level 2 | ||
1. | HRA not provided with Interlock as per Regulation 4488, section 8.2 (I.8) | 1. | Instrumentation and controls are not clearly identified Regulation 4488, section 8.1 (I.8) |
2. | Interlock does not require manual reset Regulation 4488, section 8.5 (I.8) | 2. | Scram buttons do not require manual reset Regulation 4488, section 8.6 (I.8) |
3. | Safety interlocks not independently wired Regulation 4488, sections 8.3 and 8.4 (I.8) | 3. | Not all HRA entrances are equipped with warning lights as per Regulation 4488, section 9.1 (I.9) |
4. | All safety interlocks are not fail safe Regulation 4488, section 8.4 (I.8) | 4. | Not all HRA are equipped with audible warning devices which activate for 15 seconds Regulation 4488, section 9.2 (I.9) |
5. | Scram buttons not located in HRA Regulation 4488, section 8.6 (I.8) | 5. | HRA barriers/pathways are not identified per Regulation 4483, section 8.0; Regulation 4488, section 9.3 (D.801, I.9) |
Operating Procedures
Severity Level 1 | Severity Level 2 | ||
1. | Particle accelerator not unsecured from unauthorized use Regulation 4488, section 10.1 (I.10) | ||
2. | Bypass of safety interlock not authorized by Radiation Safety Officer and/or Radiation Safety Committee or not recorded and posted as per Regulation 4488, section 10.5 (I.10) | 2. | Safety interlocks are used as routine "off" switch Regulation 4488, section 10.2 (I.10) |
3. | Electrical circuit diagram not maintained and available Regulation 4488, section 10.4 (I.10) | ||
4. | Current operating and emergency procedures are not kept at control panel Regulation 4488, section 10.6 (I.10) | ||
5. | Record of quarterly safety device operability check are not available Regulation 4488, section 10.3 (I.10) | ||
Radiation Monitoring
Severity Level 1 | Severity Level 2 | ||
1. | Radiation protection survey not performed and/or documented by an approved person following operation or facility changes Regulation 4488, section 11.2 (I.11) | 1. | Portable monitoring equipment not available, operable and calibrated Regulation 4488, section 11.1 (I.11) |
2. | Radiation levels not continuously monitored in all HRAs as per Regulation 4488, section 11.3 (I.11) | 2. | All area monitors are not calibrated annually Regulation 4488, section 11.4 (I.11) |
3. | Periodic surveys of airborne particulates are not performed as per Regulation 4488, section 11.5 (I.11) | ||
4. | Periodic Smear Surveys are not conducted for contamination as per Regulation 4488, section 11.6 (I.11) | ||
5. | All area surveys are not performed according to proper written procedures as per Regulation 4488, section 11.7 (I.11) | ||
6. | Current records of all surveys and tests were not available at facility Regulation 4488, section 11.8 (I.11) | ||
Ventilation Systems
Severity Level 1 | |||
1. | Means are not provided to ensure compliance with Part D airborne concentrations Regulation 4488, section 12.1 (I.12) | ||
2. | Airborne concentrations in excess of Part D limits were discharged to an uncontrolled area Regulation 4488, section 12.2 (I.12) | ||
Notice and Reports to Workers
Posting of Notices
Severity Level 1 | Severity Level 2 | ||
1. | Regulations 4483 and 4489 of DRCR not posted, Regulation 4489, section 2.1 (J.11) | ||
2. | Facility radiation permit/document not posted Regulation 4489, section 2.1 (J.11) | ||
3. | Radiation operating procedures not posted Regulation 4489, sections 2.1 - 2.3 (J.11) | ||
4. | Notice of violation for radiological working conditions not posted Regulation 4489, section 2.1 (J.11) | ||
5. | Notice to employees, Agency Form X not posted Regulation 4489, section 2.3 (J.11) | ||
6. | Violations not posted within five (5) days nor remain posted for at least (5) days Regulation 4489, section 2.4 (J.11) | ||
7. | Documents do not appear in sufficient number of conspicuous places Regulation 4489, section 2.5 (J.11) | ||
Instructions to Workers
Severity Level 1 | Severity Level 2 | ||
1. | Workers were not instructed to report any conditions that could cause unnecessary exposure to radiation Regulation 4489, section 3.1 (J.12) | 1. | Workers were not kept informed of radiation sources Regulation 4489, section 3.1 (J.12) |
2. | Workers were not instructed about warnings for unusual occurrence or malfunction that may involve exposure to radiation Regulation 4489, section 3.1 (J.12) | 2. | Workers were not instructed of health protection problems associated with radiation exposure Regulation 4489, section 3.1. (J.12) |
3. | Workers were not advised of radiation exposure reports pursuant to Regulation 4489, sections 3.1.1 - 3.1.6; 4.0 (J.12) | 3. | Workers were not instructed to observe applicable parts of DRCR Regulation 4489, section 3.1 - 3.1.3 (J.12) |
Notifications
Severity Level 1 | Severity Level 2 | ||
1. | Written report of specified Radiation exposure data are not given to the worker Regulation 4489, section 4.1 (J.13) | 1. | Workers are not advised annually of their radiation exposure pursuant to Regulation 4489, section 3.1 (J.13) |
2. | Workers are not furnished a radiation exposure report within 30 days after the licensee becomes informed of exposure or termination of employment Regulation 4489, section 4.3 (J.13) | 2. | As required pursuant to D.405 D.1202, D.1203, or D.1204, exposed individuals are not provided a report of their radiation exposure as per Regulation 4489, section 4.4 (J.13) |
Representatives of Registrant/Workers
Severity Level 1 | |||
1. | Agency not afforded opportunity to inspect equipment and activities Regulation 4489, section 5.1 (J.14) | ||
2. | Agency not permitted to consult with workers privately Regulation 4489, section 5.2 (J.14) | ||
3. | Worker authorized representative not given opportunity to accompany agency during inspection of physical working conditions Regulation 4489, section 5.3 (J.14) | ||
4. | Worker representative does not meet qualifications set forth in routine radiation Regulation 4489, section 5.4 (J.14) | ||
5. | Different facility/worker representatives not permitted to accompany agency on inspection Regulation 4489, section 5.5 (J.14) | ||
6. | Mutually agreed upon "outside" individual(s) was not permitted to accompany agency inspectors Regulation 4489, section 5.6 (J.14) | ||
7. | Worker(s) was not allowed to privately consult with the Agency inspector about perceived radiological condition Regulation 4489, section 6.2 (J.15) | ||
8. | A worker has been discharged or discriminated against for filing a radiological complaint on behalf of himself or others Regulation 4489, section 6.3 (J.16) | ||
Therapeutic Radiation Machines
Severity Level 1 | Severity Level 2 | ||
1. | All non-compliance with the requirements of 4492 (Part X) are Severity Level 1 violations. | ||
1.1 This Part establishes requirements, for which the registrant is responsible, for use of therapeutic radiation machines. The provisions of this Part are in addition to, and not in substitution for, other applicable provisions of the regulations.
1.2 The use of therapeutic radiation machines shall be by, or under the supervision of, a licensed practitioner of the healing arts who meets the training/experience criteria established by Part X, Section 3.0, as applicable.
As used in this Regulation, the following definitions apply:
"Absorbed dose (D)" means the mean energy imparted by ionizing radiation to matter. Absorbed dose is determined as the quotient of dE by dM, where dE is the mean energy imparted by ionizing radiation to matter of mass dM. The SI unit of absorbed dose is joule per kilogram and the special name of the unit of absorbed dose is the gray (Gy). The previously used special unit of absorbed dose (rad) is being replaced by the gray.
"Conventional Simulator" means any x-ray system designed to reproduce the geometric conditions of the radiation therapy equipment.
"Detector" (See "Radiation detector").
"Dose monitor unit (DMU)" means a unit response from the beam monitoring system from which the absorbed dose can be calculated.
"Electronic brachytherapy" means a method of radiation therapy where an electrically generated source of ionizing radiation is placed in or near the tumor or target tissue to deliver therapeutic radiation dosage.
"Electronic brachytherapy device" means the system used to produce and deliver therapeutic radiation including the x-ray tube, the control mechanism, the cooling system, and the power source.
"Electronic brachytherapy source" means the x-ray tube component used in an electronic brachytherapy device.
"External beam radiation therapy" means therapeutic irradiation in which the source of radiation is at a distance from the body.
"Field‑flattening filter" means a filter used to homogenize the absorbed dose rate over the radiation field.
"Filter" means material placed in the useful beam to change beam quality in therapeutic radiation machines subject to 6.4.
"Gray (Gy)" means the SI unit of absorbed dose, kerma, and specific energy imparted equal to 1 joule per kilogram. The previous unit of absorbed dose (rad) is being replaced by the gray [1 Gy=100 rad].
"Half‑value layer (HVL)" means the thickness of a specified material which attenuates X‑radiation or gamma radiation to an extent such that the air kerma rate, exposure rate or absorbed dose rate is reduced to one‑half of the value measured without the material at the same point.
"Intensity Modulated Radiation Therapy (IMRT)" means radiation therapy that uses non-uniform radiation beam intensities which have been determined by various computer-based optimization techniques.
"Interlock" means a device preventing the start or continued operation of equipment unless certain predetermined conditions prevail.
"Interruption of irradiation" means the stopping of irradiation with the possibility of continuing irradiation without resetting of operating conditions at the control panel.
"Irradiation" means the exposure of a living being or matter to ionizing radiation.
"Isocenter" means the center of the sphere through which the useful beam axis passes while the gantry moves through its full range of motions.
"Kilovolt (kV) [kilo electron volt (keV)]" means the energy equal to that acquired by a particle with one electron charge in passing through a potential difference of one thousand volts in a vacuum. [Note: current convention is to use kV for photons and keV for electrons.]
"Lead equivalent" means the thickness of the material in question affording the same attenuation, under specified conditions, as lead.
"Leakage radiation" means radiation emanating from the radiation therapy system except for the useful beam.
"Licensed Practitioner" means an individual licensed to practice medicine, dentistry, dental hygiene, podiatry, chiropractic, advanced practice nursing, or osteopathy in this State.
"Light field" means the area illuminated by light, simulating the radiation field.
"mA" means milliampere.
"Megavolt (MV) [mega electron volt (MeV)]" means the energy equal to that acquired by a particle with one electron charge in passing through a potential difference of one million volts in a vacuum. [Note: current convention is to use MV for photons and MeV for electrons.]
"Misadministration" means an event that meets the criteria in 5.2.
"Mobile Electronic Brachytherapy Service" means transportation of an electronic brachytherapy device to provide electronic brachytherapy at an address that is not the address of record.
"Monitor unit (MU)" (See "Dose monitor unit").
"Moving beam radiation therapy" means radiation therapy with any planned displacement of radiation field or patient relative to each other, or with any planned change of absorbed dose distribution. It includes arc, skip, conformal, intensity modulation and rotational therapy.
"Nominal treatment distance" means:
a. For electron irradiation, the distance from the scattering foil, virtual source, or exit window of the electron beam to the entrance surface of the irradiated object along the central axis of the useful beam.
b. For x‑ray irradiation, the virtual source or target to isocenter distance along the central axis of the useful beam. For non‑isocentric equipment, this distance shall be that specified by the manufacturer.
"Patient" means an individual subjected to machine produced radiation for the purposes of medical therapy.
"Peak tube potential" means the maximum value of the potential difference across the x‑ray tube during an exposure.
"Periodic quality assurance check" means a procedure which is performed to ensure that a previous parameter or condition continues to be valid.
"Phantom" means an object behaving in essentially the same manner as tissue, with respect to absorption or scattering of the ionizing radiation in question.
"Practical range of electrons" corresponds to classical electron range where the only remaining contribution to dose is from bremsstrahlung x‑rays. A further explanation may be found in "Clinical Electron Beam Dosimetry: Report of AAPM Radiation Therapy Committee Task Group 25" [Medical Physics 18(1): 73‑109, Jan/Feb. 1991] and ICRU Report 35, "Radiation Dosimetry: Electron Beams with Energies Between 1 and 50 MeV", International Commission on Radiation Units and Measurements, September 15, 1984.
"Prescribed dose" means the total dose and dose per fraction as documented in the written directive. The prescribed dose is a estimation from measured data from a specified therapeutic machine using assumptions that are clinically acceptable for that treatment technique and historically consistent with the clinical calculations previously used for patients treated with the same clinical technique.
"Primary dose monitoring system" means a system which will monitor the useful beam during irradiation and which will terminate irradiation when a pre‑selected number of dose monitor units have been delivered.
"Primary protective barrier" (see "Protective barrier").
"Protective barrier" means a barrier of radiation absorbing material(s) used to reduce radiation exposure. The types of protective barriers are as follows:
a. "Primary protective barrier" means the material, excluding filters, placed in the useful beam.
b. "Secondary protective barrier" means the material which attenuates stray radiation.
“Qualified expert” means an individual who has demonstrated to the satisfaction of the Agency that such individual possesses the knowledge and training to measure ionizing radiation, to evaluate safety techniques, and to advise regarding radiation protection needs, for example, individuals certified in the appropriate field by the American Board of Radiology, or the American Board of Health Physics, or the American Board of Medical Physics, or those having equivalent qualifications. With reference to the calibration of radiation therapy equipment, an individual, in addition to the above qualifications, must be qualified in accordance with 4492.3.3 and 4492.3.4.
"Qualified Medical Physicist" means an individual qualified in accordance with 3.3 Part X, subsections 3.2 and 3.4.
"Radiation detector" means a device which, in the presence of radiation provides, by either direct or indirect means, a signal or other indication suitable for use in measuring one or more quantities of incident radiation.
"Radiation field" (see "Useful beam").
"Radiation head" means the structure from which the useful beam emerges.
"Redundant beam monitoring system" means a combination of two independent dose monitoring systems in which each system is designed to terminate irradiation in accordance with a pre‑selected number of dose monitor units.
"Scattered radiation" means ionizing radiation emitted by interaction of ionizing radiation with matter, the interaction being accompanied by a change in direction of the radiation. Scattered primary radiation means that scattered radiation which has been deviated in direction only by materials irradiated by the useful beam.
"Secondary dose monitoring system" means a system which will terminate irradiation in the event of failure of the primary dose monitoring system.
"Secondary protective barrier" (see "Protective barrier").
"Shadow tray" means a device attached to the radiation head to support auxiliary beam blocking material.
"Shutter" means a device attached to the tube housing assembly which can totally intercept the useful beam and which has a lead equivalency not less than that of the tube housing assembly.
"Sievert (Sv)" means the SI unit of dose equivalent. The unit of dose equivalent is the joule per kilogram. The previous unit of dose equivalent (rem) is being replaced by the sievert. [1 Sv=100 rem.]
"Simulator (radiation therapy simulation system)" means any x‑ray system intended for localizing the volume to be exposed during radiation therapy and establishing the position and size of the therapeutic irradiation field. [See: Conventional Simulator and Virtual Simulator.]
"Source" means the region and/or material from which the radiation emanates.
"Source‑skin distance (SSD)" (see "Target‑skin distance").
"Stationary beam radiation therapy" means radiation therapy without displacement of one or more mechanical axes relative to the patient during irradiation.
"Stray radiation" means the sum of leakage and scattered radiation.
"Target" means that part of an x‑ray tube or accelerator onto which a beam of accelerated particles is directed to produce ionizing radiation or other particles.
"Target‑skin distance (TSD)" means the distance measured along the beam axis from the center of the front surface of the x‑ray target and/or electron virtual source to the surface of the irradiated object or patient.
"Tenth‑value layer (TVL)" means the thickness of a specified material which attenuates X‑radiation or gamma radiation to an extent such that the air kerma rate, exposure rate, or absorbed dose rate is reduced to one‑tenth of the value measured without the material at the same point.
"Termination of irradiation" means the stopping of irradiation in a fashion which will not permit continuance of irradiation without the resetting of operating conditions at the control panel.
"Therapeutic radiation machine" means x‑ray or electron‑producing equipment designed and used for external beam radiation therapy. For the purpose of these regulations, devices used to administer electronic brachytherapy shall also be considered therapeutic radiation machines.
"Tube" means an x‑ray tube, unless otherwise specified.
"Tube housing assembly" means the tube housing with tube installed. It includes high‑voltage and/or filament transformers and other appropriate elements when such are contained within the tube housing.
"Useful beam" means the radiation emanating from the tube housing port or the radiation head and passing through the aperture of the beam limiting device when the exposure controls are in a mode to cause the therapeutic radiation machine to produce radiation.
"Virtual Simulator" means a computed tomography (CT) unit used in conjunction with relevant software which recreates the treatment machine; and that allows import, manipulation, display, and storage of images from CT and/or other imaging modalities.
"Virtual source" means a point from which radiation appears to originate.
"Wedge filter" means a filter which effects continuous change in transmission over all or a part of the useful beam.
"Written directive" means an order in writing for the administration of radiation to a specific patient or human research subject, as specified in 5.1.
"X‑ray tube" means any electron tube which is designed to be used primarily for the production of x‑rays.
3.1 Administrative Controls. The registrant shall be responsible for directing the operation of the therapeutic radiation machines that have been registered with the Agency. The registrant or the registrant's agent shall ensure that the requirements of Part X are met in the operation of the therapeutic radiation machine(s).
3.2 A therapeutic radiation machine that does not meet the provisions of these regulations shall not be used for irradiation of patients.
3.3 Training for Therapeutic Radiation Machine Authorized Users. The registrant for any therapeutic radiation machine subject to 6.0 or 7.0 shall require the authorized user to be a physician who:
3.3.1 Is certified in:
3.3.1.1 Radiation oncology or therapeutic radiology by the American Board of Radiology or Radiology (combined diagnostic and therapeutic radiology program) by the American Board of Radiology prior to 1976; or
3.3.1.2 Radiation oncology by the American Osteopathic Board of Radiology; or
3.3.1.3 Radiology, with specialization in radiotherapy, as a British "Fellow of the Faculty of Radiology" or "Fellow of the Royal College of Radiology"; or
3.3.1.4 Therapeutic radiology by the Canadian Royal College of Physicians and Surgeons; or
3.3.2 Is in the active practice of therapeutic radiology, and has completed 200 hours of instruction in basic radiation techniques applicable to the use of an external beam radiation therapy unit, five hundred (500) hours of supervised work experience, and a minimum of three (3) years of supervised clinical experience.
3.3.2.1 To satisfy the requirement for instruction, the classroom and laboratory training shall include:
3.3.2.1.1 Radiation physics and instrumentation;
3.3.2.1.2 Radiation protection;
3.3.2.1.3 Mathematics pertaining to the use and measurement of ionization radiation; and
3.3.2.1.4 Radiation biology.
3.3.2.2 To satisfy the requirement for supervised work experience, training shall be under the supervision of an authorized user and shall include:
3.3.2.2.1 Review of the full calibration measurements and periodic quality assurance checks;
3.3.2.2.2 Evaluation of prepared treatment plans and calculation of treatment times/patient treatment settings;
3.3.2.2.3 Using administrative controls to prevent misadministrations;
3.3.2.2.4 Implementing emergency procedures to be followed in the event of the abnormal operation of an external beam radiation therapy unit or console; and
3.3.2.2.5 Checking and using radiation survey meters.
3.3.2.3 To satisfy the requirement for a period of supervised clinical experience, training shall include one (1) year in a formal training program approved by the Residency Review Committee for Radiology of the Accreditation Council for Graduate Medical Education or the Committee on Postdoctoral Training of the American Osteopathic Association and an additional two (2) years of clinical experience in therapeutic radiology under the supervision of an authorized user. The supervised clinical experience shall include:
3.3.2.3.1 Examining individuals and reviewing their case histories to determine their suitability for external beam radiation therapy treatment, and any limitations/contraindications;
3.3.2.3.2 Selecting proper dose and how it is to be administered;
3.3.2.3.3 Calculating the therapeutic radiation machine doses and collaborating with the authorized user in the review of patients' progress and consideration of the need to modify originally prescribed doses and/or treatment plans as warranted by patients' reaction to radiation; and
3.3.2.3.4 Post‑administration follow‑up and review of case histories.
3.3.3 Notwithstanding the requirements of 3.31 and 3.3.2 the registrant for any therapeutic radiation machine subject to 6.0 may also submit the training of the prospective authorized user physician for Agency review on a case‑by‑case basis.
3.3.4 A physician shall not act as an authorized user for any therapeutic radiation machine until such time as said physician's training has been reviewed and approved by the Agency.] 3*/
3.4 Training for Qualified Medical Physicist. The registrant for any therapeutic radiation machine subject to 6.0 or 7.0 shall require the Qualified Medical Physicist to:
3.4.1 Be registered with the Agency, under the provisions of Part B of these regulations, as a provider of radiation services in the area of calibration and compliance surveys of external beam radiation therapy units; and
3.4.2 Be certified by the American Board of Radiology in:
3.4.2.1 Therapeutic medical physics; or
3.4.2.2 Diagnostic medical physics; or
3.4.2.3 Nuclear medical physics; or
3.4.2.4 Radiological physics; or
3.4.2.5 Be certified by the American Board of Medical Physics in Radiation Oncology Physics; or
3.4.2.6 Be certified by the Canadian College of Medical Physics; or
3.4.2.7 Hold a master's or doctor's degree in physics, medical physics, other physical science, engineering, or applied mathematics from an accredited college or university, and have completed one (1) year of full time training in medical physics and an additional year of full time work experience under the supervision of a Qualified Medical Physicist at a medical institution. This training and work experience shall be conducted in clinical radiation facilities that provide high-energy external beam radiation therapy (photons and electrons with energies greater than or equal to one MV/one MeV). To meet this requirement, the individual shall have performed the tasks listed in 4.1, 6.16, 7.20 and 6.16, 7.21 under the supervision of a Qualified Medical Physicist during the year of work experience.
3.5 Qualifications of Operators.
3.5.1 Individuals who will be operating a therapeutic radiation machine for medical use shall be American Registry of Radiologic Technologists (ARRT) Registered Radiation Therapy Technologists. Individuals who are not ARRT Registered Radiation Therapy Technologists shall submit evidence that they have satisfactorily completed a radiation therapy technologist training program that complies with the requirements of the Joint Review Committee on Education in Radiologic Technology.5/
3.5.2 The names and training of all personnel currently operating a therapeutic radiation machine shall be kept on file at the facility. Information on former operators shall be retained for a period of at least two (2) years beyond the last date they were authorized to operate a therapeutic radiation machine at that facility.]
3.6 Written safety procedures and rules shall be developed by a Qualified Medical Physicist and shall be available in the control area of a therapeutic radiation machine, including any restrictions required for the safe operation of the particular therapeutic radiation machine. The operator shall be able to demonstrate familiarity with these rules.
3.7 Individuals shall not be exposed to the useful beam except for medical therapy purposes and unless such exposure has been ordered in writing by a therapeutic radiation machine authorized user. This provision specifically prohibits deliberate exposure of an individual for training, demonstration or other non‑healing‑arts purposes.
3.8 Visiting Authorized User. Notwithstanding the provisions of 3.7 a registrant may permit any physician to act as a visiting authorized user under the term of the registrant's Certificate of Registration for up to sixty (60) days per calendar year under the following conditions:
3.8.1 The visiting authorized user has the prior written permission of the registrant's management and, if the use occurs on behalf of an institution, the institution's Radiation Safety Committee (where applicable); and
3.8.2 The visiting authorized user meets the requirements established for authorized user(s) in 3.3.1 and 3.3.2 and
3.8.3 The registrant shall maintain copies of the written permission required in 3.8.1 and documentation that the visiting authorized user met the requirements of 3.8.2 for five (5) years from the date of the last visit.
3.9 All individuals associated with the operation of a therapeutic radiation machine shall be instructed in and shall comply with the provisions of the registrant's quality management program. In addition to the requirements of Part X, these individuals are also subject to the requirements of Parts D.1201, D.1502 and D.2104 of these regulations.
3.10 Information and Maintenance Record and Associated Information. The registrant shall maintain the following information in a separate file or package for each therapeutic radiation machine, for inspection by the Agency:
3.10.1 Report of acceptance testing;
3.10.2 Records of all surveys, calibrations, and periodic quality assurance checks of the therapeutic radiation machine required by Part X, as well as the name(s) of person(s) who performed such activities;
3.10.3 Records of maintenance and/or modifications performed on the therapeutic radiation machine, as well as the name(s) of person(s) who performed such services;
3.10.4 Signature of person authorizing the return of therapeutic radiation machine to clinical use after service, repair, or upgrade.
3.11 Records Retention. All records required by Part X shall be retained until disposal is authorized by the Agency unless another retention period is specifically authorized in Part X. All required records shall be retained in an active file from at least the time of generation until the next Agency inspection. Any required record generated prior to the last Agency inspection may be microfilmed or otherwise archived as long as a complete copy of said record can be retrieved until such time as the Agency authorizes final disposal.
4.1 Protection Surveys.
4.1.1 The registrant shall ensure that radiation protection surveys of all new facilities, and existing facilities not previously surveyed are performed with an operable radiation measurement survey instrument calibrated in accordance with 8.0. The radiation protection survey shall be performed by, or under the direction of, a Qualified Medical Physicist or a qualified expert and shall verify that, with the therapeutic radiation machine in a "BEAM‑ON" condition, with the largest clinically available treatment field and with a scattering phantom in the useful beam of radiation:
4.1.1.1 Radiation levels in restricted areas are not likely to cause personnel exposures in excess of the limits specified in Part D.1201a.of these regulations; and
4.1.1.2 Radiation levels in unrestricted areas do not exceed the limits specified in Parts D.1301a. and D.1301b. of these regulations.
4.1.2.1 After making any change in the treatment room shielding;
4.1.2.2 After making any change in the location of the therapeutic radiation machine within the treatment room;
4.1.2.3 After relocating the therapeutic radiation machine; or
4.1.2.4 Before using the therapeutic radiation machine in a manner that could result in increased radiation levels in areas outside the external beam radiation therapy treatment room.
4.1.3 The survey record shall indicate all instances where the facility, in the opinion of the Qualified Medical Physicist or a Qualified Expert, is in violation of applicable regulations. The survey record shall also include: the date of the measurements; the reason the survey is required; the manufacturer's name; model number and serial number of the therapeutic radiation machine; the instrument(s) used to measure radiation levels; a plan of the areas surrounding the treatment room that were surveyed; the measured dose rate at several points in each area expressed in microsieverts or millirems per hour; the calculated maximum level of radiation over a period of one (1) week for each restricted and unrestricted area; and the signature of the individual responsible for conducting the survey;
4.1.4 If the results of the surveys required by 4.1.1.1 or 4.1.1.2 indicate any radiation levels in excess of the respective limit specified in 4.1.1.1 the registrant shall lock the control in the "OFF" position and not use the unit:
4.1.4.1 Except as may be necessary to repair, replace, or test the therapeutic radiation machine, the therapeutic radiation machine shielding, or the treatment room shielding; or
4.1.4.2 Until the registrant has received a specific exemption from the Agency.
4.2 Modification of Radiation Therapy Unit or Room Before Beginning a Treatment Program. If the survey required by 4.1.1 indicates that an individual in an unrestricted area may be exposed to levels of radiation greater than those permitted by Parts D.1301a. and D.1301b. of these regulations, before beginning the treatment program the registrant shall:
4.2.1 Either equip the unit with beam direction interlocks or add additional radiation shielding to ensure compliance with Parts D.1301a. and D.1301b. of these regulations;
4.2.2 Perform the survey required by 4.1 again; and
4.2.3 Include in the report required by 4.4 the results of the initial survey, a description of the modification made to comply with 4.2.1 and the results of the second survey; or
4.2.4 Request and receive a registration amendment under Part D.1301c.of these regulations that authorizes radiation levels in unrestricted areas greater than those permitted by Parts D.1301a. and D.1301b. of these regulations.
4.3 Dosimetry Equipment.
4.3.1 The registrant shall have a calibrated dosimetry system available for use. The system shall have been calibrated by the National Institute for Standards and Technology (NIST) or by an American Association of Physicists in Medicine (AAPM) Accredited Dosimetry Calibration Laboratory (ADCL). The calibration shall have been performed within the previous twenty-four (24) months and after any servicing that may have affected system calibration. An independent survey shall be conducted by a qualified expert or Qualified Medical Physicist other than the person performing the original survey prior to the equipment being used except as described in X.4a.iv.
4.3.1.1 For beams with energies greater than 1 MV (1 MeV), the dosimetry system shall have been calibrated for Cobalt‑60;
4.3.1.2 For beams with energies equal to or less than 1 MV (1 MeV), the dosimetry system shall have been calibrated at an energy (energy range) appropriate for the radiation being measured;
4.3.2 The registrant shall have available for use a dosimetry system for quality assurance check measurements. To meet this requirement, the system may be compared with a system that has been calibrated in accordance with 4.3.1. This comparison shall have been performed within the previous twelve (12) months and after each servicing that may have affected system calibration. The quality assurance check system may be the same system used to meet the requirement in 4.3.1
4.4 Reports of External Beam Radiation Therapy Surveys and Measurements. The registrant for any therapeutic radiation machine subject to 6.0 or 7.0 shall furnish a copy of the records required in 4.1 and 4.2 to the Agency within thirty (30) days following completion of the action that initiated the record requirement.
Each registrant or applicant subject to 6.0, 7.0 or 11.0 shall develop, implement, and maintain a quality management program to provide high confidence that radiation will be administered as directed by the authorized user.
5.1 Scope and Applicability. Each registrant or applicant subject to subsection 6.0, 7.0 or 11.0 shall develop, implement, and maintain a quality management program to provide high confidence that radiation will be administered as directed by the authorized user. The quality management program shall address, as a minimum, the following specific objectives:
5.1.1 Written Directives:
5.1.1.1 A written directive must be dated and signed by an authorized user prior to the administration of radiation.
If because of the patient’s condition, a delay in the order to provide a written revision to an existing written directive would jeopardize the patient’s health, an oral revision to an existing written directive will be acceptable, provided that the oral revision is documented as soon as possible in writing in the patient’s record and a revised written directive is signed by an authorized user within 48 hours of the oral revision.
5.1.1.2 The written directive must contain the patient or human research subject’s name, the type and energy of the beam, the total dose, dose per fraction, treatment site, and number of fractions.
5.1.1.3 A written revision to an existing written directive may be made provided that the revision is dated and signed by an authorized user prior to the administration of the therapeutic radiation machine dose, or the next fractional dose.
5.1.1.4 The registrant shall retain a copy of the written directive for three (3) years.
5.1.2 Procedures for Administrations. The registrant shall develop, implement, and maintain written procedures to provide high confidence that:
5.1.2.1 Prior to the administration of each course of radiation treatments, the patient’s or human research subject’s identity is verified by more than one method as the individual named in the written directive;
5.1.2.2 Each administration is in accordance with the written directive;
5.1.2.3 Therapeutic radiation machine final plans of treatment and related calculations are in accordance with the respective written directives by:
5.1.2.3.1 Checking both manual and computer generated dose calculations to verify they are correct and in accordance with the written directive; and
5.1.2.3.2 Verifying that any computer-generated calculations are correctly transferred into the consoles of authorized therapeutic medical units;
5.1.2.4 Any unintended deviation from the written directive is identified, evaluated and appropriate action is taken; and
5.1.2.5 The registrant retains a copy of the procedures for administrations for the duration of the registration.
5.2 Reports and Notifications of Misadministrations.
5.2.1 A registrant shall report any event resulting from intervention of a patient or human research subject in which the administration of therapeutic radiation machine radiation results, or will result in, unintended permanent functional damage to an organ or a physiological system as determined by a physician.
5.2.2 Other than events that result from intervention by a patient or human research subject, a registrant shall report any event in which the administration of a therapeutic radiation machine therapy dose:
5.2.2.1 Involves the wrong patient, wrong treatment modality, or wrong treatment site; or
5.2.2.2 The calculated weekly administered dose differs from the weekly prescribed dose by more than thirty percent (30%); or
5.2.2.3 The calculated total administered dose differs from the total prescribed dose by more than twenty percent (20%) of the total prescribed dose;
5.2.4.1 The registrant’s name;
5.2.4.2 The name of the prescribing physician;
5.2.4.3 A brief description of the event;
5.2.4.4 Why the event occurred;
5.2.4.5 The effect, if any, on the individuals(s) who received the administration;
5.2.4.6 Actions, if any, that have been taken, or are planned, to prevent recurrence;
5.2.4.7 Certification that the registrant notified the individual (or the individual’s responsible relative or guardian), and if not, why not.
5.2.5 The report shall not contain the individual’s name or any other information that could lead to the identification of the individual.
5.2.6 The registrant shall provide notification of the event to the referring physician and also notify the individual who is the subject of the misadministration no later than twenty-four (24) hours after its discovery, unless the referring physician personally informs the registrant either that he or she will inform the individual or that, based on medical judgment, telling the individual would be harmful. The registrant is not required to notify the individual without first consulting the referring physician. If the referring physician or the affected individual cannot be reached within twenty-four (24) hours, the registrant shall notify the individual as soon as possible thereafter. The registrant may not delay any appropriate medical care for the individual, including any necessary remedial care as a result of the misadministration, because of any delay in notification. To meet the requirements of this paragraph, the notification of the individual who is the subject of the misadministration may be made instead to that individual’s responsible relative or guardian. If a verbal notification is made, the registrant shall inform the individual, or appropriate responsible relative or guardian, that a written description of the event can be obtained from the registrant upon request. The registrant shall provide such a written description if requested.
5.2.7 Aside from the notification requirement, nothing in this section affects any rights or duties of registrants and physicians in relation to each other, to individuals affected by the misadministration, or to that individual’s responsible relatives or guardians.
5.2.8 The registrant shall retain a record of a misadministration in accordance with 5.3. A copy of the record required shall be provided to the referring physician if other than the registrant within fifteen (15) days after discovery of the misadministration.
5.3 Records of Misadministrations. A registrant shall retain a record of misadministrations reported in accordance with 5.2 for three (3) years. The record must contain the following:
5.3.1 The registrant’s name and the names of the individuals involved;
5.3.2 The social security number or other identification number, if one has been assigned, of the individual who is the subject of the misadministration;
5.3.3 A brief description of the event; why it occurred; the effect, if any, on the individual;
5.3.4 The actions, if any, taken or planned to prevent recurrence; and
5.3.5 Whether the registrant notified the individual (or the individual’s responsible relative or guardian) and, if not, whether such failure to notify was based on guidance from the referring physician.
6.1 Leakage Radiation. When the x‑ray tube is operated at its maximum rated tube current for the maximum kV, the leakage air kerma rate shall not exceed the value specified at the distance specified for that classification of therapeutic radiation machine:
6.1.1 5‑50 kV Systems. The leakage air kerma rate measured at any position 5 centimeters from the tube housing assembly shall not exceed 1 mGy (100 mrad) in any one hour.
6.1.2 >50 and <500 kV Systems. The leakage air kerma rate measured at a distance of 1 meter from the target in any direction shall not exceed 1 cGy (1 rad) in any 1 hour. This air kerma rate measurement may be averaged over areas no larger than one hundred square centimeters (100 cm2). In addition, the air kerma rate at a distance of 5 centimeters from the surface of the tube housing assembly shall not exceed 30 cGy (30 rad) per hour.
6.1.3 For each therapeutic radiation machine, the registrant shall determine, or obtain from the manufacturer, the leakage radiation existing at the positions specified in 6.1.1 and 6.1.2 for the specified operating conditions. Records on leakage radiation measurements shall be maintained at the installation for inspection by the Agency.
6.2 Permanent Beam Limiting Devices. Permanent diaphragms or cones used for limiting the useful beam shall provide at least the same degree of attenuation as required for the tube housing assembly.
6.3 Adjustable or Removable Beam Limiting Devices.
6.3.1 All adjustable or removable beam limiting devices, diaphragms, cones or blocks shall not transmit more than 5 percent of the useful beam for the most penetrating beam used;
6.3.2 When adjustable beam limiting devices are used, the position and shape of the radiation field shall be indicated by a light beam.
6.4 Filter System. The filter system shall be so designed that:
6.4.1 Filters can not be accidentally displaced at any possible tube orientation;
6.4.2 For equipment installed after July 10, 2002 an interlock system prevents irradiation if the proper filter is not in place;
6.4.3 The air kerma rate escaping from the filter slot shall not exceed 1 cGy (1 rad) per hour at one (1) meter under any operating conditions; and
6.4.4 Each filter shall be marked as to its material of construction and its thickness.
6.5 Tube Housing.
6.5.1 The x‑ray tube shall be so mounted that it can not accidentally turn or slide with respect to the housing aperture; and
6.5.2 The tube housing assembly shall be capable of being immobilized for stationary portal treatments.
6.5.3 Source Marking. The tube housing assembly shall be so marked that it is possible to determine the location of the source to within 5 millimeters, and such marking shall be readily accessible for use during calibration procedures.
6.6 Beam Block. Contact therapy tube housing assemblies shall have a removable shield of material, equivalent in attenuation to 0.5 millimeters of lead at 100 kV, which can be positioned over the entire useful beam exit port during periods when the beam is not in use.
6.7 Timer. A suitable irradiation control device shall be provided to terminate the irradiation after a pre‑set time interval.
6.7.1 A timer with a display shall be provided at the treatment control panel. The timer shall have a pre‑set time selector and an elapsed time or time remaining indicator;
6.7.2 The timer shall be a cumulative timer that activates with an indication of "BEAM‑ON" and retains its reading after irradiation is interrupted or terminated. After irradiation is terminated and before irradiation can be reinitiated, it shall be necessary to reset the elapsed time indicator;
6.7.3 The timer shall terminate irradiation when a pre‑selected time has elapsed, if any dose monitoring system present has not previously terminated irradiation;
6.7.4 The timer shall permit accurate pre‑setting and determination of exposure times as short as 1 second;
6.7.5 The timer shall not permit an exposure if set at zero;
6.7.6 The timer shall not activate until the shutter is opened when irradiation is controlled by a shutter mechanism unless calibration includes a timer error correction to compensate for mechanical lag; and
6.7.7 Timer shall be accurate to within 1 percent of the selected value or 1 second, whichever is greater.
6.8 Control Panel Functions. The control panel, in addition to the displays required by other provisions in 6.0 shall have:
6.8.1 An indication of whether electrical power is available at the control panel and if activation of the x‑ray tube is possible;
6.8.2 An indication of whether x‑rays are being produced;
6.8.3 A means for indicating x‑ray tube potential and current;
6.8.4 The means for terminating an exposure at any time;
6.8.5 A locking device which will prevent unauthorized use of the therapeutic radiation machine; and
6.8.6 For therapeutic radiation machines manufactured after July 10, 2002, a positive display of specific filter(s) in the beam.
6.9 Multiple Tubes. When a control panel may energize more than one x‑ray tube:
6.9.1 It shall be possible to activate only one x‑ray tube at any time;
6.9.2 There shall be an indication at the control panel identifying which x‑ray tube is activated; and
6.9.3 There shall be an indication at the tube housing assembly when that tube is energized.
6.10 Target‑to‑Skin Distance (TSD). There shall be a means of determining the central axis TSD to within one (1) centimeter and of reproducing this measurement to within two (2) millimeters thereafter.
6.11 Shutters. Unless it is possible to bring the x‑ray output to the prescribed exposure parameters within 5 seconds after the x‑ray "ON" switch is energized, the beam shall be attenuated by a shutter having a lead equivalency not less than that of the tube housing assembly. In addition, after the unit is at operating parameters, the shutter shall be controlled by the operator from the control panel. An indication of shutter position shall appear at the control panel.
6.12 Low Filtration X‑ray Tubes. Each therapeutic radiation machine equipped with a beryllium or other low‑filtration window shall be clearly labeled as such upon the tube housing assembly and shall be provided with a permanent warning device on the control panel that is activated when no additional filtration is present, to indicate that the dose rate is very high.
6.13 Facility Design Requirements for Therapeutic Radiation Machines Capable of Operating in the Range 50 kV to 500 kV. In addition to shielding adequate to meet requirements of 9.0, the treatment room shall meet the following design requirements:
6.13.1 Aural Communication. Provision shall be made for continuous two‑way aural communication between the patient and the operator at the control panel;
6.13.2 Viewing Systems. Provision shall be made to permit continuous observation of the patient during irradiation and the viewing system shall be so located that the operator can observe the patient from the control panel. The therapeutic radiation machine shall not be used for patient irradiation unless at least one viewing system is operational.
6.14 Additional Requirements. Treatment rooms that contain a therapeutic radiation machine capable of operating above 150 kV shall meet the following additional requirements:
6.14.1 All protective barriers shall be fixed except for entrance doors or beam interceptors;
6.14.2 The control panel shall be located outside the treatment room or in a totally enclosed booth, which has a ceiling, inside the room;
6.14.3 Interlocks shall be provided such that all entrance doors, including doors to any interior booths, shall be closed before treatment can be initiated or continued. If the radiation beam is interrupted by any door opening, it shall not be possible to restore the machine to operation without closing the door and reinitiating irradiation by manual action at the control panel; and
6.14.4 When any door referred to in 6.14.3 is opened while the x‑ray tube is activated, the air kerma rate at a distance of 1 meter from the source shall be reduced to less than 1 mGy (100 mrad) per hour.
6.15 Full Calibration Measurements.
6.15.1 Full calibration of a therapeutic radiation machine subject to 6.0 shall be performed by, or under the direct supervision of, a Qualified Medical Physicist:
6.15.1.1 Before the first medical use following installation or reinstallation of the therapeutic radiation machine;
6.15.1.2 At intervals not exceeding one (1) year; and
6.15.1.3 Before medical use under the following conditions:
6.15.1.3.1 Whenever quality assurance check measurements indicate that the radiation output differs by more than five percent (5%) from the value obtained at the last full calibration and the difference cannot be reconciled; and
6.15.1.3.2 Following any component replacement, major repair, or modification of components that could significantly affect the characteristics of the radiation beam.
6.15.1.4 Notwithstanding the requirements of 6.15.1.3:
6.15.1.4.1 Full calibration of therapeutic radiation machines with multi‑energy capabilities is required only for those modes and/or energies that are not within their acceptable range; and
6.15.1.4.2 If the repair, replacement or modification does not affect all energies, full calibration shall be performed on the affected energy that is in most frequent clinical use at the facility. The remaining energies may be validated with quality assurance check procedures against the criteria in 6.15.1.4.1
6.16 Periodic Quality Assurance Checks.
6.16.1 Periodic quality assurance checks shall be performed on therapeutic radiation machines subject to 6.0 which are capable of operation at greater than or equal to 50 kV.
6.16.2 To satisfy the requirement of 6.16.1 quality assurance checks shall meet the following requirements:
6.16.2.1 The registrant shall perform quality assurance checks in accordance with written procedures established by the Qualified Medical Physicist; and
6.16.2.2 The quality assurance check procedures shall specify the frequency at which tests or measurements are to be performed. The quality assurance check procedures shall specify that the quality assurance check shall be performed during the calibration specified in 6.15.1. The acceptable tolerance for each parameter measured in the quality assurance check, when compared to the value for that parameter determined in the calibration specified in 6.15.1 shall be stated.
6.16.3 The cause for a parameter exceeding a tolerance set by the Qualified Medical Physicist shall be investigated and corrected before the system is used for patient irradiation;
6.16.4 Whenever a quality assurance check indicates a significant change in the operating characteristics of a system, as specified in the Qualified Medical Physicist's quality assurance check procedures, the system shall be recalibrated as required in 6.15.1
6.16.5 The registrant shall use the dosimetry system described in 4.3.2 to make the quality assurance check required in 6.15.2;
6.16.6 The registrant shall have the Qualified Medical Physicist review and sign the results of each radiation output quality assurance check within thirty (30) days of the date that the check was performed;
6.16.7 The registrant shall ensure that safety quality assurance checks of therapeutic radiation machines subject to 6.0 are performed at intervals not to exceed thirty (30) days;
6.16.8 Notwithstanding the requirements of 6.16.6 and 6.16.7, the registrant shall ensure that no therapeutic radiation machine is used to administer radiation to humans unless the quality assurance checks required by 6.16.6 and 6.16.7 have been performed within the thirty (30) day period immediately prior to said administration;
6.16.9 To satisfy the requirement of 6.16.7 safety quality assurance checks shall ensure proper operation of:
6.16.9.1 Electrical interlocks at each external beam radiation therapy room entrance;
6.16.9.2 The "BEAM‑ON" and termination switches;
6.16.9.3 Beam condition indicator lights on the access door(s), control console, and in the radiation therapy room;
6.16.9.4 Viewing systems;
6.16.9.5 If applicable, electrically operated treatment room doors from inside and outside the treatment room;
6.16.10 The registrant shall maintain a record of each quality assurance check required by 6.16.1 and 6.16.7 for 3 years. The record shall include: the date of the quality assurance check; the manufacturer's name, model number, and serial number of the therapeutic radiation machine; the manufacturer's name; model number and serial number for the instrument(s) used to measure the radiation output of the therapeutic radiation machine; and the signature of the individual who performed the periodic quality assurance check
6.17 Operating Procedures.
6.17.1 The therapeutic radiation machine shall not be used for irradiation of patients unless the requirements of 6.15 and 6.16 have been met;
6.17.2 Therapeutic radiation machines shall not be left unattended unless secured pursuant to 6.7.5
6.17.3 When a patient must be held in position for radiation therapy, mechanical supporting or restraining devices shall be used;
6.17.4 The tube housing assembly shall not be held by an individual during operation unless the assembly is designed to require such holding and the peak tube potential of the system does not exceed 50 kV. In such cases, the holder shall wear protective gloves and apron of not less than 0.5 millimeters lead equivalency at 100 kV;
6.17.5 A copy of the current operating and emergency procedures shall be maintained at the therapeutic radiation machine control console; and
6.17.6 No individual other than the patient shall be in the treatment room during exposures from therapeutic radiation machines operating above 150 kV. At energies less than or equal to 150 kV, any individual, other than the patient, in the treatment room shall be protected by a barrier sufficient to meet the requirements of Part D.1201 of these regulations.
6.18 Possession of Survey Instrument(s). Each facility location authorized to use a therapeutic radiation machine in accordance with 6.0 shall possess appropriately calibrated portable monitoring equipment.
6.18.1 As a minimum, such equipment shall include a portable radiation measurement survey instrument capable of measuring dose rates over the range 10 μSv (1 mrem) per hour to 10 mSv (1000 mrem) per hour. The survey instrument(s) shall be operable and calibrated in accordance with 8.0
7.1 Possession of Survey Instrument(s). Each facility location authorized to use a therapeutic radiation machine in accordance with 7.0 shall possess appropriately calibrated portable monitoring equipment.
7.1.1 As a minimum, such equipment shall include a portable radiation measurement survey instrument capable of measuring dose rates over the range 10 µSv (1 mrem) per hour to 10 mSv (1000 mrem) per hour. The survey instrument(s) shall be operable and calibrated in accordance with 8.0
7.2 Leakage Radiation Outside the Maximum Useful Beam in Photon and Electron Modes.
7.2.1 The absorbed dose due to leakage radiation (excluding neutrons) at any point outside the maximum sized useful beam, but within a circular plane of radius two (2) meters which is perpendicular to and centered on the central axis of the useful beam at the nominal treatment distance (i.e. patient plane), shall not exceed a maximum of 0.2 percent and an average of 0.1 percent of the absorbed dose on the central axis of the beam at the nominal treatment distance. Measurements shall be averaged over an area not exceeding one hundred square centimeters (100 cm2) at a minimum of sixteen (16) points uniformly distributed in the plane;
7.2.2 Except for the area defined in 7.2.1 the absorbed dose due to leakage radiation (excluding neutrons) at 1 meter from the electron path between the electron source and the target or electron window shall not exceed 0.5 percent of the absorbed dose on the central axis of the beam at the nominal treatment distance. Measurements shall be averaged over an area not exceeding one hundred square centimeters(100 cm2);
7.2.3 For equipment manufactured after July 10, 2002 the neutron absorbed dose outside the useful beam shall be in compliance with International Electrotechnical Commission (IEC) Document 60601‑2‑1 (most current revision); and
7.2.4 For each therapeutic radiation machine, the registrant shall determine, or obtain from the manufacturer, the leakage radiation existing at the positions specified in 7.2.1 through 7.2.3 for the specified operating conditions. Records on leakage radiation measurements shall be maintained at the installation for inspection by the Agency.
7.3 Leakage Radiation Through Beam Limiting Devices.
7.3.1 Photon Radiation. All adjustable or interchangeable beam limiting devices shall attenuate the useful beam such that at the nominal treatment distance, the maximum absorbed dose anywhere in the area shielded by the beam limiting device(s) shall not exceed 2 percent of the maximum absorbed dose on the central axis of the useful beam measured in a 100 cm2 radiation field, or maximum available field size if less than 100 cm2;
7.3.2 Electron Radiation. All adjustable or interchangeable electron applicators shall attenuate the radiation, including but not limited to photon radiation generated by electrons incident on the beam limiting device and electron applicator and other parts of the radiation head, such that the absorbed dose in a plane perpendicular to the central axis of the useful beam at the nominal treatment distance shall not exceed:
7.3.2.1 A maximum of two percent (2%) and average of 0.5 percent of the absorbed dose on the central axis of the useful beam at the nominal treatment distance. This limit shall apply beyond a line seven (7) centimeters outside the periphery of the useful beam; and
7.3.2.2 A maximum of ten percent (10%) of the absorbed dose on the central axis of the useful beam at the nominal treatment distance. This limit shall apply beyond a line two (2) centimeters outside the periphery of the useful beam.
7.3.2.3 Measurement of Leakage Radiation.
7.3.2.3.1 Photon Radiation. Measurements of leakage radiation through the beam limiting devices shall be made with the beam limiting devices closed and any residual aperture blocked by at least two (2) tenth value layers of suitable absorbing material. In the case of overlapping beam limiting devices, the leakage radiation through each set shall be measured independently at the depth of maximum dose. Measurements shall be made using a radiation detector of area not exceeding ten square centimeters (10 cm2);
7.3.2.3.2 Electron Radiation. Measurements of leakage radiation through the electron applicators shall be made with the electron beam directed into the air and using a radiation detector of area up to but not exceeding one (1) square centimeter suitably protected against radiation which has been scattered from material beyond the radiation detector. Measurements shall be made using one (1) centimeter of water equivalent build up material.
7.4 Filters/Wedges.
7.4.1 Each wedge filter that is removable from the system shall be clearly marked with an identification number. For removable wedge filters, the nominal wedge angle shall appear on the wedge or wedge tray (if permanently mounted to the tray). If the wedge or wedge tray is significantly damaged, the wedge transmission factor shall be redetermined;
7.4.2 If the absorbed dose rate information required by 7.9 relates exclusively to operation with a field flattening filter or beam scattering foil in place, such foil or filter shall be removable only by the use of tools;
7.4.3 For equipment manufactured after July 10, 2002 which utilizes wedge filters, interchangeable field flattening filters, or interchangeable beam scattering foils:
7.4.3.1 Irradiation shall not be possible until a selection of a filter or a positive selection to use "no filter" has been made at the treatment control panel, either manually or automatically;
7.4.3.2 An interlock system shall be provided to prevent irradiation if the filter selected is not in the correct position;
7.4.3.3 A display shall be provided at the treatment control panel showing the wedge filter(s), interchangeable field flattening filter(s), and/or interchangeable beam scattering foil(s) in use; and
7.4.3.4 An interlock shall be provided to prevent irradiation if any filter and/or beam scattering foil selection operation carried out in the treatment room does not agree with the filter and/or beam scattering foil selection operation carried out at the treatment control panel.
7.5 Stray Radiation in the Useful Beam. For equipment manufactured after July 10, 2002, the registrant shall determine during acceptance testing, or obtain from the manufacturer, data sufficient to ensure that x‑ray stray radiation in the useful electron beam, absorbed dose at the surface during x‑ray irradiation and stray neutron radiation in the useful x‑ray beam are in compliance with International Electrotechnical Commission (IEC) Document 60601‑2‑1 (most current revision).
7.6 Beam Monitors. All therapeutic radiation machines subject to 7.0 shall be provided with redundant beam monitoring systems. The sensors for these systems shall be fixed in the useful beam during treatment to indicate the dose monitor unit rate.
7.6.1 Equipment manufactured after July 10, 2002 shall be provided with at least two (2) independently powered integrating dose meters. Alternatively, common elements may be used if the production of radiation is terminated upon failure of any common element.
7.6.2 Equipment manufactured on or before July 10, 2002 shall be provided with at least one (1) radiation detector. This detector shall be incorporated into a useful beam monitoring system;
7.6.3 The detector and the system into which that detector is incorporated shall meet the following requirements:
7.6.3.1 Each detector shall be removable only with tools and, if movable, shall be interlocked to prevent incorrect positioning;
7.6.3.2 Each detector shall form part of a beam monitoring system from whose readings in dose monitor units the absorbed dose at a reference point can be calculated;
7.6.3.3 Each beam monitoring system shall be capable of independently monitoring, interrupting, and terminating irradiation; and
7.6.3.4 For equipment manufactured after July 10, 2002, the design of the beam monitoring systems shall ensure that the:
7.6.3.4.1 Malfunctioning of one system shall not affect the correct functioning of the other system(s); and
7.6.3.4.2 Failure of either system shall terminate irradiation or prevent the initiation of radiation.
7.6.3.5 Each beam monitoring system shall have a legible display at the treatment control panel. For equipment manufactured after July 10, 2002, each display shall:
7.6.3.5.1 Maintain a reading until intentionally reset;
7.6.3.5.2 Have only one scale and no electrical or mechanical scale multiplying factors;
7.6.3.5.3 Utilize a design such that increasing dose is displayed by increasing numbers; and
7.6.3.5.4 In the event of power failure, the beam monitoring information required in 7.6.3.5.3 displayed at the control panel at the time of failure shall be retrievable
7.7 Beam Symmetry.
7.7.1 A bent‑beam linear accelerator with beam flattening filter(s) subject to 7.0 shall be provided with auxiliary device(s) to monitor beam symmetry;
7.7.2 The device(s) referenced in 7.7.1 shall be able to detect field asymmetry greater than ten percent (10%); and
7.7.3 The device(s) referenced in 7.7.1 shall be configured to terminate irradiation if the specifications in 7.7.2 can not be maintained.
7.8 Selection and Display of Dose Monitor Units.
7.8.1 Irradiation shall not be possible until a new selection of a number of dose monitor units has been made at the treatment control panel;
7.8.2 The pre‑selected number of dose monitor units shall be displayed at the treatment control panel until reset manually for the next irradiation;
7.8.3 After termination of irradiation, it shall be necessary to reset the dosimeter display before subsequent treatment can be initiated; and
7.8.4 For equipment manufactured after July 10, 2002, after termination of irradiation, it shall be necessary for the operator to reset the pre‑selected dose monitor units before irradiation can be initiated.
7.9 Air Kerma Rate/Absorbed Dose Rate. For equipment manufactured after July 10, 2002, a system shall be provided from whose readings the air kerma rate or absorbed dose rate at a reference point can be calculated. [The radiation detectors specified in 7.6 may form part of this system.] In addition:
7.9.1 The dose monitor unit rate shall be displayed at the treatment control panel;
7.9.2 If the equipment can deliver under any conditions an air kerma rate or absorbed dose rate at the nominal treatment distance more than twice the maximum value specified by the manufacturer, a device shall be provided which terminates irradiation when the air kerma rate or absorbed dose rate exceeds a value twice the specified maximum. The dose rate at which the irradiation will be terminated shall be a record maintained by the registrant;
7.9.3 If the equipment can deliver under any fault condition(s) an air kerma rate or absorbed dose rate at the nominal treatment distance more than ten (10) times the maximum value specified by the manufacturer, a device shall be provided to prevent the air kerma rate or absorbed dose rate anywhere in the radiation field from exceeding twice the specified maximum value and to terminate irradiation if the excess absorbed dose at the nominal treatment distance exceeds 4 Gy (400 rad); and
7.9.4 For each therapeutic radiation machine, the registrant shall determine, or obtain from the manufacturer, the maximum value(s) specified in 7.9.2 and 7.9.3 for the specified operating conditions. Records of these maximum value(s) shall be maintained at the installation for inspection by the Agency.
7.10 Termination of Irradiation by the Beam Monitoring System or Systems During Stationary Beam Radiation Therapy.
7.10.1 Each primary system shall terminate irradiation when the pre‑selected number of dose monitor units has been detected by the system;
7.10.2 If the original design of the equipment included a secondary dose monitoring system, that system shall be capable of terminating irradiation when not more than fifteen percent (15%) or forty (40) dose monitor units above the pre‑selected number of dose monitor units set at the control panel has been detected by the secondary dose monitoring system; and
7.10.3 For equipment manufactured after July 10, 2002, an indicator on the control panel shall show which monitoring system has terminated irradiation.
7.11 Termination of Irradiation. It shall be possible to terminate irradiation and equipment movement or go from an interruption condition to termination condition at any time from the operator's position at the treatment control panel.
7.12 Interruption of Irradiation. If a therapeutic radiation machine has an interrupt mode, it shall be possible to interrupt irradiation and equipment movements at any time from the treatment control panel. Following an interruption it shall be possible to restart irradiation by operator action without any reselection of operating conditions. If any change is made of a pre‑selected value during an interruption, irradiation and equipment movements shall be automatically terminated.
7.13 Timer. A suitable irradiation control device shall be provided to terminate the irradiation after a pre‑set time interval.
7.13.1 A timer shall be provided which has a display at the treatment control panel. The timer shall have a pre‑set time selector and an elapsed time indicator;
7.13.2 The timer shall be a cumulative timer that activates with an indication of "BEAM‑ON" and retains its reading after irradiation is interrupted or terminated. After irradiation is terminated and before irradiation can be reinitiated, it shall be necessary to reset the elapsed time indicator;
7.13.3 The timer shall terminate irradiation when a pre‑selected time has elapsed, if the dose monitoring systems have not previously terminated irradiation.
7.14 Selection of Radiation Type. Equipment capable of both x‑ray therapy and electron therapy shall meet the following additional requirements:
7.14.1 Irradiation shall not be possible until a selection of radiation type (x‑rays or electrons) has been made at the treatment control panel;
7.14.2 The radiation type selected shall be displayed at the treatment control panel before and during irradiation;
7.14.3 An interlock system shall be provided to ensure that the equipment can principally emit only the radiation type that has been selected;
7.14.4 An interlock system shall be provided to prevent irradiation with x‑rays, except to obtain an image, when electron applicators are fitted;
7.14.5 An interlock system shall be provided to prevent irradiation with electrons when accessories specific for x‑ray therapy are fitted; and
7.14.6 An interlock system shall be provided to prevent irradiation if any selected operations carried out in the treatment room do not agree with the selected operations carried out at the treatment control panel.
7.15 Selection of Energy. Equipment capable of generating radiation beams of different energies shall meet the following requirements:
7.15.1 Irradiation shall not be possible until a selection of energy has been made at the treatment control panel;
7.15.2 The nominal energy value selected shall be displayed at the treatment control panel until reset manually for the next irradiation. After termination of irradiation, it shall be necessary to reset the nominal energy value selected before subsequent treatment can be initiated;
7.15.3 Irradiation shall not be possible until the appropriate flattening filter or scattering foil for the selected energy is in its proper location; and
7.15.4 For equipment manufactured after July 10, 2002, the selection of energy shall be in compliance with International Electrotechnical Commission (IEC) Document 60601‑2‑1 (most current revision).
7.16 Selection of Stationary Beam Radiation Therapy or Moving Beam Radiation Therapy. Therapeutic radiation machines capable of both stationary beam radiation therapy and moving beam radiation therapy shall meet the following requirements:
7.16.1 Irradiation shall not be possible until a selection of stationary beam radiation therapy or moving beam radiation therapy has been made at the treatment control panel;
7.16.2 The mode of operation shall be displayed at the treatment control panel;
7.16.3 An interlock system shall be provided to ensure that the equipment can operate only in the mode that has been selected;
7.16.4 An interlock system shall be provided to prevent irradiation if any selected parameter in the treatment room does not agree with the selected parameter at the treatment control panel;
7.16.5 Moving beam radiation therapy shall be controlled to obtain the selected relationships between incremental dose monitor units and incremental movement. For equipment manufactured after July 10, 2002:
7.16.5.1 An interlock system shall be provided to terminate irradiation if the number of dose monitor units delivered in any 10 degrees of rotation or 1 cm of linear motion differs by more than twenty percent (20%) from the selected value;
7.16.5.2 Where angle terminates the irradiation in moving beam radiation therapy, the dose monitor units delivered shall differ by less than five percent (5%) from the dose monitor unit value selected;
7.16.5.3 An interlock shall be provided to prevent motion of more than five (5) degrees or one (1) cm beyond the selected limits during moving beam radiation therapy;
7.16.5.4 An interlock shall be provided to require that a selection of direction be made at the treatment control panel in all units which are capable of both clockwise and counter‑clockwise moving beam radiation therapy.
7.16.5.5 Moving beam radiation therapy shall be controlled with both primary position sensors and secondary position sensors to obtain the selected relationships between incremental dose monitor units and incremental movement.
7.16.6 Where the beam monitor system terminates the irradiation in moving beam radiation therapy, the termination of irradiation shall be as required by 7.10 and
7.16.7 For equipment manufactured after July 10, 2002, an interlock system shall be provided to terminate irradiation if movement:
7.16.7.1 Occurs during stationary beam radiation therapy; or
7.16.7.2 Does not start or stops during moving beam radiation therapy unless such stoppage is a pre‑planned function.
7.17 Facility Design Requirements for Therapeutic Radiation Machines Operating above 500 kV. In addition to shielding adequate to meet requirements of 9.0 the following design requirements are made:
7.17.1 Protective Barriers. All protective barriers shall be fixed, except for access doors to the treatment room or movable beam interceptors;
7.17.2 Control Panel. In addition to other requirements specified in Part X, the control panel shall also:
7.17.2.1 Be located outside the treatment room;
7.17.2.2 Provide an indication of whether electrical power is available at the control panel and if activation of the radiation is possible;
7.17.2.3 Provide an indication of whether radiation is being produced; and
7.17.2.4 Include an access control (locking) device that will prevent unauthorized use of the therapeutic radiation machine;
7.17.3 Viewing Systems. Windows, mirrors, closed‑circuit television or an equivalent viewing system shall be provided to permit continuous observation of the patient following positioning and during irradiation and shall be so located that the operator may observe the patient from the treatment control panel. The therapeutic radiation machine shall not be used for patient irradiation unless at least one viewing system is operational;
7.17.4 Aural Communications. Provision shall be made for continuous two‑way aural communication between the patient and the operator at the control panel. The therapeutic radiation machine shall not be used for irradiation of patients unless continuous two‑way aural communication is possible;
7.17.5 Room Entrances. Treatment room entrances shall be provided with warning lights in a readily observable position near the outside of all access doors, which will indicate when the useful beam is "ON" and when it is "OFF";
7.17.6 Entrance Interlocks. Interlocks shall be provided such that all access controls are activated before treatment can be initiated or continued. If the radiation beam is interrupted by any access control, it shall not be possible to restore the machine to operation without resetting the access control and reinitiating irradiation by manual action at the control panel;
7.17.7 Beam Interceptor Interlocks. If the shielding material in any protective barrier requires the presence of a beam interceptor to ensure compliance with Parts D.1301a. and D.1301b. of these regulations, interlocks shall be provided to prevent the production of radiation, unless the beam interceptor is in place, whenever the useful beam is directed at the designated barrier(s);
7.17.8 Emergency Cutoff Switches. At least 1 emergency power cutoff switch shall be located in the radiation therapy room and shall terminate all equipment electrical power including radiation and mechanical motion. This switch is in addition to the termination switch required by 7.11. All emergency power cutoff switches shall include a manual reset so that the therapeutic radiation machine cannot be restarted from the unit's control console without resetting the emergency cutoff switch;
7.17.9 Safety Interlocks. All safety interlocks shall be designed so that any defect or component failure in the safety interlock system prevents or terminates operation of the therapeutic radiation machine; and
7.17.10 Surveys for Residual Radiation. Surveys for residual activity shall be conducted on all therapeutic radiation machines capable of generating photon and electron energies above 10 MV prior to machining, removing, or working on therapeutic radiation machine components which may have become activated due to photo‑neutron production.
7.18 Qualified Medical Physicist Support.
7.18.1 The services of a Qualified Medical Physicist shall be required in facilities having therapeutic radiation machines with energies of 500 kV and above. The Qualified Medical Physicist shall be responsible for:
7.18.1.1 Full calibration(s) required by 7.20 and protection surveys required by 4.1
7.18.1.2 Supervision and review of dosimetry;
7.18.1.3 Beam data acquisition and transfer for computerized dosimetry, and supervision of its use;
7.18.1.4 Quality assurance, including quality assurance check review required by 7.21.4
7.18.1.5 Consultation with the authorized user in treatment planning, as needed; and
7.18.1.6 Perform calculations/assessments regarding misadministrations.
7.18.2 If the Qualified Medical Physicist is not a full‑time employee of the registrant, the operating procedures required by 7.19 shall also specifically address how the Qualified Medical Physicist is to be contacted for problems or emergencies, as well as the specific actions, if any, to be taken until the Qualified Medical Physicist can be contacted.
7.19 Operating Procedures.
7.19.1 No individual, other than the patient, shall be in the treatment room during treatment or during any irradiation for testing or calibration purposes;
7.19.2 Therapeutic radiation machines shall not be made available for medical use unless the requirements of 4.1, 7.20 and 7.21 have been met;
7.19.3 Therapeutic radiation machines, when not in operation, shall be secured to prevent unauthorized use;
7.19.4 When adjustable beam limiting devices are used, the position and shape of the radiation field shall be indicated by a light field.
7.19.5 If a patient must be held in position during treatment, mechanical supporting or restraining devices shall be used; and
7.19.6 A copy of the current operating and emergency procedures shall be maintained at the therapeutic radiation machine control console.
7.20 Acceptance Testing, Commissioning and Full Calibration Measurements.
7.20.1 Acceptance testing, commissioning and full calibration of a therapeutic radiation machine subject to 7.0 shall be performed by, or under the direct supervision of, a Qualified Medical Physicist.
7.20.2 Acceptance testing and commissioning shall be performed in accordance with "AAPM Code of Practice for Radiotherapy Accelerators: AAPM Report No. 47", prepared by Radiation Therapy Task Group 45 and the manufacturer’s contractual specifications. Acceptance testing and commissioning shall be conducted before the first medical use following installation or reinstallation of the therapeutic radiation machine.
7.20.3 Full calibration shall include measurement of all applicable parameters required by Table II of "Comprehensive QA for Radiation Oncology: Report of AAPM Radiation Therapy: AAPM Report No. 46," prepared by Committee Task Group 40 and shall be performed in accordance with "AAPM Code of Practice for Radiotherapy Accelerators: AAPM Report No. 47" prepared by Radiation Therapy Task Group 45. Although it shall not be necessary to complete all elements of a full calibration at the same time, all applicable parameters (for all energies) shall be completed at intervals not exceeding twelve (12) calendar months, unless a more frequent interval is required in Table II.
7.20.4 The Qualified Medical Physicist shall perform all elements of a full calibration necessary to determine that all parameters are within acceptable limits:
7.20.4.1 Whenever quality assurance check measurements indicate that the radiation output differs by more than five percent (5%) from the value obtained at the last full calibration and the difference cannot be reconciled. Therapeutic radiation machines with multi‑energy and/or multi‑mode capabilities shall only require measurements for those modes and/or energies that are not within their acceptable range; and
7.20.4.2 Following any component replacement, major repair, or modification of components that could significantly affect the characteristics of the radiation beam. If the repair, replacement or modification does not affect all modes and/or energies, measurements shall be performed on the effected mode/energy that is in most frequent clinical use at the facility. The remaining energies/modes may be validated with quality assurance check procedures against the criteria in 7.20.4.1
7.20.5 The registrant shall maintain a record of each calibration in an auditable form for the duration of the registration. The record shall include: the date of the calibration; the manufacturer's name, model number and serial number for the therapeutic radiation machine; the model numbers and serial numbers of the instruments used to calibrate the therapeutic radiation machine; and the signature of the Qualified Medical Physicist responsible for performing the calibration.
7.21 Periodic Quality Assurance Checks.
7.21.1 Periodic quality assurance checks shall be performed on all therapeutic radiation machines subject to 7.0 at intervals not to exceed those specified in "Comprehensive QA for Radiation Oncology: AAPM Report No. 46,” prepared by AAPM Radiation Therapy Committee Task Group 40;
7.21.2 To satisfy the requirement of 7.21.1, quality assurance checks shall include determination of central axis radiation output and a representative sampling of periodic quality assurance checks contained in "Comprehensive QA for Radiation Oncology: AAPM Report No. 46 prepared by Radiation Therapy Committee Task Group 40. Representative sampling shall include all applicable referenced periodic quality assurance checks in an interval not to exceed twelve (12) consecutive calendar months;
7.21.3 The registrant shall use a dosimetry system that has been intercompared within the previous twelve (12) months with the dosimetry system described in 4.3.1 to make the periodic quality assurance checks required in 7.21.2
7.21.4 The registrant shall perform periodic quality assurance checks required by 7.21.1 in accordance with procedures established by the Qualified Medical Physicist;
7.21.5 The registrant shall review the results of each periodic radiation output check according to the following procedures:
7.21.5.1 The authorized user and Qualified Medical Physicist shall be immediately notified if any parameter is not within its acceptable tolerance. The therapeutic radiation machine shall not be made available for subsequent medical use until the Qualified Medical Physicist has determined that all parameters are within their acceptable tolerances;
7.21.5.2 If all quality assurance check parameters appear to be within their acceptable range, the quality assurance check shall be reviewed and signed by either the authorized user or Qualified Medical Physicist within 3 treatment days; and
7.21.5.3 The Qualified Medical Physicist shall review and sign the results of each radiation output quality assurance check at intervals not to exceed thirty (30) days.
7.21.6 Therapeutic radiation machines subject to 7.0 shall have applicable safety quality assurance checks listed in "Comprehensive QA for Radiation Oncology: AAPM Report No. 46," prepared by AAPM Radiation Therapy Committee Task Group 40 performed at intervals not to exceed 1 week;
7.21.7 To satisfy the requirement of 7.21.6 safety quality assurance checks shall ensure proper operation of:
7.21.7.1 Electrical interlocks at each external beam radiation therapy room entrance;
7.21.7.2 Proper operation of the "BEAM‑ON", interrupt and termination switches;
7.21.7.3 Beam condition indicator lights on the access doors, control console, and in the radiation therapy room;
7.21.7.4 Viewing systems;
7.21.7.5 Electrically operated treatment room door(s) from inside and outside the treatment room;
7.21.7.6 At least one emergency power cutoff switch. If more than one emergency power cutoff switch is installed and not all switches are tested at once, each switch shall be tested on a rotating basis. Safety quality assurance checks of the emergency power cutoff switches may be conducted at the end of the treatment day in order to minimize possible stability problems with the therapeutic radiation machine.
7.21.7.7 The registrant shall promptly repair any system identified in 7.21.7 that is not operating properly; and
7.21.8 The registrant shall maintain a record of each quality assurance check required by 7.21.1 and 7.21.7 for three (3) years.
7.21.8.1 The record shall include: the date of the quality assurance check; the manufacturer's name, model number, and serial number of the therapeutic radiation machine; the manufacturer's name, model number and serial number for the instrument(s) used to measure the radiation output of the therapeutic radiation machine; and the signature of the individual who performed the periodic quality assurance check.
7.21.9 Quality Assurance Checks for IMRT shall:
7.21.9.1 Include commissioning and testing of the treatment planning and delivery systems, routine quality assurance of the delivery system, and patient-specific validation of treatment plans;7/ and
7.21.9.2 Be performed in accordance with "Guidance document on delivery, treatment planning, and clinical implementation of IMRT: Report of the IMRT subcommittee of the AAPM radiation therapy committee: AAPM Report No. 82”; and
7.21.9.3 Be performed in accordance with the manufacturer’s contractual specifications.
8.1 The registrant shall ensure that the survey instruments used to show compliance with 4492 (Part X) have been calibrated before first use, at intervals not to exceed twelve (12) months, and following repair. The registrant shall check each survey instrument for consistent response with a dedicated check source before each use. The licensee is not required to keep records of these checks.
8.2 To satisfy the requirements of 8.1 the registrant shall:
8.2.1 Calibrate all required scale readings up to 10 mSv (1000 mrem) per hour with an appropriate radiation source that is traceable to the National Institute of Standards and Technology (NIST);
8.2.2 Calibrate at least two (2) points on each scale to be calibrated. These points should be at approximately 1/3 and 2/3 of full-scale; and
8.3 To satisfy the requirements of 8.2 the registrant shall:
8.3.1 Consider a point as calibrated if the indicated dose rate differs from the calculated dose rate by not more than 10 percent; and
8.3.2 Consider a point as calibrated if the indicated dose rate differs from the calculated dose rate by not more than 20 percent if a correction factor or graph is conspicuously attached to the instrument.
8.4 The registrant shall retain a record of each calibration required in 8.1 for three (3) years. The record shall include:
8.4.1 A description of the calibration procedure; and
8.4.2 A description of the source used and the certified dose rates from the source, and the rates indicated by the instrument being calibrated, the correction factors deduced from the calibration data, the signature of the individual who performed the calibration, and the date of calibration.
8.5 The registrant may obtain the services of individuals licensed by the Agency, the US Nuclear Regulatory Commission or an Agreement State to perform calibrations of survey instruments. Records of calibrations that contain information required by 8.4 shall be maintained by the registrant.
9.1 Each therapeutic radiation machine subject to 6.0 or 7.0 shall be provided with such primary and/or secondary barriers as are necessary to ensure compliance with Parts D.1201 and D.1301 of these regulations.
9.2 Facility design information for all new installations of a therapeutic radiation machine or installations of a therapeutic radiation machine of higher energy into a room not previously approved for that energy shall be submitted for Agency approval prior to actual installation of the therapeutic radiation machine. The minimum facility design information that must be submitted is contained in Appendix A to (Part X).
10.1 Quality assurance for a conventional or virtual simulator shall include acceptance testing and periodic verification of system performance; and
10.2 Be performed in accordance with "Comprehensive QA for Radiation Oncology: Report of AAPM Radiation Therapy Committee Task Group No.40: AAPM Report No. 46” for a conventional simulator; or
10.3 Be performed in accordance with “Quality assurance for computed tomography simulators and the computed tomography-simulation process: Report of the AAPM Radiation Therapy Committee Task Group No. 66: AAPM Report No. 83” for a virtual simulator.
11.1 Applicability. Electronic brachytherapy devices shall be subject to the requirements of 11.0 and shall be exempt for the requirements of 6.0.
11.1.1 An electronic brachytherapy device that does not meet the requirements of 11.0 shall not be used for irradiation of patients; and
11.1.2 An electronic brachytherapy device shall only be utilized for human use applications specifically approved by the U.S. Food and Drug Administration (FDA) unless participating in a research study approved by the registrant’s Institutional Review Board (IRB).
11.2 Possession of Survey Instrument(s). Each facility location authorized to use an electronic brachytherapy device in accordance with 11.0 shall possess appropriately calibrated portable monitoring equipment. As a minimum, such equipment shall include a portable radiation measurement survey instrument capable of measuring dose rates over the range 10 µSv (1 mrem) per hour to 10 mSv (1000 mrem) per hour. The survey instrument(s) shall be operable and calibrated in accordance with 8.0 for the applicable electronic brachytherapy source energy.
11.3 Facility Design Requirements for Electronic Brachytherapy Devices. In addition to shielding adequate to meet requirements of 9.0 the treatment room shall meet the following design requirements:
11.3.1 If applicable, provision shall be made to prevent simultaneous operation of more than one therapeutic radiation machine in a treatment room.
11.3.2 Access to the treatment room shall be controlled by a door at each entrance.
11.3.3 Each treatment room shall have provisions to permit continuous aural communication and visual observation of the patient from the treatment control panel during irradiation. The electronic brachytherapy device shall not be used for patient irradiation unless the patient can be observed.
11.3.4 For electronic brachytherapy devices capable of operating below 50 kV, radiation shielding for the staff in the treatment room shall be available, either as a portable shield and/or as localized shielded material around the treatment site.
11.3.5 For electronic brachytherapy devices capable of operating at greater than 150 kV: 8/
11.3.5.1 The control panel shall be located outside the treatment room; and
11.3.5.2 Electrical interlocks shall be provided for all door(s) to the treatment room that will:
11.3.5.2.1 Prevent the operator from initiating the treatment cycle unless each treatment room entrance door is closed;
11.3.5.2.2 Cause the source to be shielded when an entrance door is opened; and
11.3.5.2.3 Prevent the source from being exposed following an interlock interruption until all treatment room entrance doors are closed and the source on-off control is reset at the console.
11.4 Electrical Safety for Electronic Brachytherapy Devices.
11.4.1 The high voltage transformer shall be electrically isolated to prevent electrical and magnetic interference with the surrounding environment and ancillary equipment.
11.4.2 The high voltage transformer shall be isolated from personnel (e.g., operator) and the environment by a protective housing that can only be accessed through a cover requiring a tool for access or with electrical interlocks to prevent operation while open.
11.4.3 The high voltage transformer shall have appropriate safety labels warning personnel of potential electrical shock and/or heat related injuries.
11.4.4 Equipment manufactured after shall be in compliance with the most current revision of the following International Electrotechnical Commission (IEC) Documents:
11.4.4.1 IEC 60601-1:1998+A1+A2:1995;
11.4.4.2 IEC 60601-1-2:2001;
11.4.4.3 IEC 60601-2-8:1999; and
11.4.4.4 IEC 60601-2-17:2004.
11.5 Control Panel Functions. The control panel, in addition to the displays required by other provisions in 11.0 shall:
11.5.1 Provide an indication of whether electrical power is available at the control panel and if activation of the electronic brachytherapy source is possible;
11.5.2 Provide an indication of whether x‑rays are being produced;
11.5.3 Provide a means for indicating electronic brachytherapy source potential and current;
11.5.4 Provide the means for terminating an exposure at any time; and
11.5.5 Include an access control (locking) device that will prevent unauthorized use of the electronic brachytherapy device.
11.6 Timer. A suitable irradiation control device (timer) shall be provided to terminate the irradiation after a pre-set time interval or integrated charge on a dosimeter-based monitor.
11.6.1 A timer shall be provided at the treatment control panel. The timer shall indicate planed setting and the time elapsed or remaining;
11.6.2 The timer shall not permit an exposure if set at zero;
11.6.3 The timer shall be a cumulative device that activates with an indication of "BEAM-ON" and retains its reading after irradiation is interrupted or terminated. After irradiation is terminated and before irradiation can be reinitiated, it shall be necessary to reset the elapsed time indicator;
11.6.4 The timer shall terminate irradiation when a pre-selected time has elapsed, if any dose monitoring system has not previously terminated irradiation.
11.6.5 The timer shall permit setting of exposure times as short as 0.1 second; and
11.6.6 The timer shall be accurate to within one (1) percent of the selected value or 0.1 second, whichever is greater.
11.7 Qualified Medical Physicist Support.
11.7.1 The services of a Qualified Medical Physicist shall be required in facilities having electronic brachytherapy devices. The Qualified Medical Physicist shall be responsible for:
11.7.1.1 Evaluation of the output from the electronic brachytherapy source;
11.7.1.2 Generation of the necessary dosimetric information;
11.7.1.3 Supervision and review of treatment calculations prior to initial treatment of any treatment site;
11.7.1.4 Establishing the periodic and day-of-use quality assurance checks and reviewing the data from those checks as required in 11.11;
11.7.1.5 Consultation with the authorized user in treatment planning, as needed; and
11.7.1.6 Performing calculations/assessments regarding patient treatments that may constitute a misadministration.
11.7.2 If the Qualified Medical Physicist is not a full-time employee of the registrant, the operating procedures required by 11.8 shall also specifically address how the Qualified Medical Physicist is to be contacted for problems or emergencies, as well as the specific actions, if any, to be taken until the Qualified Medical Physicist can be contacted.
11.8 Operating Procedures.
11.8.1 Only individuals approved by the authorized user, Radiation Safety Officer, or Qualified Medical Physicist shall be present in the treatment room during treatment;
11.8.2 Electronic brachytherapy devices shall not be made available for medical use unless the requirements of 4.1, 11.9 and 11.10 have been met;
11.8.3 The electronic brachytherapy device shall be inoperable, either by hardware or password, when unattended by qualified staff or service personnel;
11.8.4 During operation, the electronic brachytherapy device operator shall monitor the position of all persons in the treatment room, and all persons entering the treatment room, to prevent entering persons from unshielded exposure from the treatment beam;
11.8.5 If a patient must be held in position during treatment, mechanical supporting or restraining devices shall be used;
11.8.6 Written procedures shall be developed, implemented, and maintained for responding to an abnormal situation. These procedures shall include:
11.8.6.1 Instructions for responding to equipment failures and the names of the individuals responsible for implementing corrective actions; and
11.8.6.2 The names and telephone numbers of the authorized users, the Qualified Medical Physicist, and the Radiation Safety Officer to be contacted if the device or console operates abnormally.
11.8.7 A copy of the current operating and emergency procedures shall be physically located at the electronic brachytherapy device control console9/;
11.8.8 Instructions shall be posted at the electronic brachytherapy device control console5/ to inform the operator of the names and telephone numbers of the authorized users, the Qualified Medical Physicist, and the Radiation Safety Officer to be contacted if the device or console operates abnormally; and
11.8.9 The Radiation Safety Officer, or his/her designee, and an authorized user shall be notified as soon as possible if the patient has a medical emergency, suffers injury or dies. The Radiation Safety Officer or the Qualified Medical Physicist shall inform the manufacturer of the event.
11.9 Safety Precautions for Electronic Brachytherapy Devices.
11.9.1 A Qualified Medical Physicist shall determine which persons in the treatment room require monitoring when the beam is energized;
11.9.2 An authorized user and a Qualified Medical Physicist shall be physically present during the initiation of all patient treatments involving the electronic brachytherapy device;
11.9.3 A Qualified Medical Physicist and either an authorized user or a physician or electronic brachytherapy device operator, under the supervision of an authorized user, who has been trained in the operation and emergency response for the electronic brachytherapy device, shall be physically present during continuation of all patient treatments involving the electronic brachytherapy device;
11.9.4 When shielding is required by 11.3.4, the electronic brachytherapy device operator shall use a survey meter to verify proper placement of the shielding immediately upon initiation of treatment. Alternatively, a Qualified Medical Physicist shall designate shield locations sufficient to meet the requirements of D.1201 of these regulations for any individual, other than the patient, in the treatment room; and
11.9.5 All personnel in the treatment room are required to remain behind shielding during treatment. A Qualified Medical Physicist shall approve any deviation from this requirement and shall designate alternative radiation safety protocols, compatible with patient safety, to provide an equivalent degree of protection.
11.10 Electronic Brachytherapy Source Calibration Measurements.
11.10.1 Calibration of the electronic brachytherapy source output for an electronic brachytherapy device subject to 11.0 shall be performed by, or under the direct supervision of, a Qualified Medical Physicist;
11.10.2 Calibration of the electronic brachytherapy source output shall be made for each electronic brachytherapy source, or after any repair affecting the x-ray beam generation, or when indicated by the electronic brachytherapy source quality assurance checks;
11.10.3 Calibration of the electronic brachytherapy source output shall utilize a dosimetry system described in 4.3;
11.10.4 Calibration of the electronic brachytherapy source output shall include, as applicable, determination of:
11.10.4.1 The output within two percent (2%) of the expected value, if applicable, or determination of the output if there is no expected value;
11.10.4.2 Timer accuracy and linearity over the typical range of use;
11.10.4.3 Proper operation of back-up exposure control devices;
11.10.4.4 Evaluation that the relative dose distribution about the source is within five percent (5%) of that expected; and
11.10.4.5 Source positioning accuracy to within one (1) millimeter within the applicator;
11.10.5 Calibration of the x-ray source output required by 11.10 through 11.10.4 shall be in accordance with current published recommendations from a recognized national professional association with expertise in electronic brachytherapy (when available). In the absence of a calibration protocol published by a national professional association, the manufacturer’s calibration protocol shall be followed.
11.10.6 The registrant shall maintain a record of each calibration in an auditable form for the duration of the registration. The record shall include: the date of the calibration; the manufacturer's name, model number and serial number for the electronic brachytherapy device and a unique identifier for it’s electronic brachytherapy source; the model numbers and serial numbers of the instrument(s) used to calibrate the electronic brachytherapy device; and the name and signature of the Qualified Medical Physicist responsible for performing the calibration.
11.11 Periodic and Day-of-Use Quality Assurance Checks for Electronic Brachytherapy Devices.
11.11.1 Quality assurance checks shall be performed on each electronic brachytherapy device subject to 11.0
11.11.1.1 At the beginning of each day of use;
11.11.1.2 Each time the device is moved to a new room or site10; and
11.11.1.3 After each x-ray tube installation.
11.11.2 The registrant shall perform periodic quality assurance checks required by 11.11.1 in accordance with procedures established by the Qualified Medical Physicist;
11.11.3 To satisfy the requirements of 11.11.1, radiation output quality assurance checks shall include as a minimum:
11.11.3.1 Verification that output of the electronic brachytherapy source falls within three percent (3%) of expected values, as appropriate for the device, as determined by:
11.11.3.1.1 Output as a function of time, or
11.11.3.1.2 Output as a function of setting on a monitor chamber.
11.11.3.2 Verification of the consistency of the dose distribution to within three percent (3%) of that found during calibration required by 11.10; and
11.11.3.3 Validation of the operation of positioning methods to ensure that the treatment dose exposes the intended location within one (1) mm; and
11.11.4 The registrant shall use a dosimetry system that has been intercompared within the previous twelve (12) months with the dosimetry system described in 4.3.1 to make the quality assurance checks required in 11.11.3;
11.11.5 The registrant shall review the results of each radiation output quality assurance check according to the following procedures:
11.11.5.1 An authorized user and Qualified Medical Physicist shall be immediately notified if any parameter is not within its acceptable tolerance. The electronic brachytherapy device shall not be made available for subsequent medical use until the Qualified Medical Physicist has determined that all parameters are within their acceptable tolerances;
11.11.5.2 If all radiation output quality assurance check parameters appear to be within their acceptable range, the quality assurance check shall be reviewed and signed by either the authorized user or Qualified Medical Physicist within two (2) days; and
11.11.5.3 The Qualified Medical Physicist shall review and sign the results of each radiation output quality assurance check at intervals not to exceed thirty (30) days.
11.11.6 To satisfy the requirements of 11.11.1, safety device quality assurance checks shall, at a minimum, assure:
11.11.6.1 Proper operation of radiation exposure indicator lights on the electronic brachytherapy device and on the control console;
11.11.6.2 Proper operation of viewing and intercom systems in each electronic brachytherapy facility, if applicable;
11.11.6.3 Proper operation of radiation monitors, if applicable;
11.11.6.4 The integrity of all cables, catheters or parts of the device that carry high voltages; and
11.11.6.5 Connecting guide tubes, transfer tubes, transfer-tube-applicator interfaces, and treatment spacers are free from any defects that interfere with proper operation.
11.11.7 If the results of the safety device quality assurance checks required in 11.11.5 indicate the malfunction of any system, a registrant shall secure the control console in the OFF position and not use the electronic brachytherapy device except as may be necessary to repair, replace, or check the malfunctioning system.
11.11.8 The registrant shall maintain a record of each quality assurance check required by 11.11.3 and 11.11.7 in an auditable form for three (3) years.
11.11.8.1 The record shall include the date of the quality assurance check; the manufacturer's name, model number and serial number for the electronic brachytherapy device; the name and signature of the individual who performed the periodic quality assurance check and the name and signature of the Qualified Medical Physicist who reviewed the quality assurance check;
11.11.8.2 For radiation output quality assurance checks required by 11.11.3 the record shall also include the unique identifier for the electronic brachytherapy source and the manufacturer's name; model number and serial number for the instrument(s) used to measure the radiation output of the electronic brachytherapy device.
11.12 Therapy-Related Computer Systems. The registrant shall perform acceptance testing on the treatment planning system of electronic brachytherapy-related computer systems in accordance with current published recommendations from a recognized national professional association with expertise in electronic brachytherapy (when available). In the absence of an acceptance testing protocol published by a national professional association, the manufacturer’s acceptance testing protocol shall be followed.
11.12.1 Acceptance testing shall be performed by, or under the direct supervision of, a Qualified Medical Physicist. At a minimum, the acceptance testing shall include, as applicable, verification of:
11.12.1.1 The source-specific input parameters required by the dose calculation algorithm;
11.12.1.2 The accuracy of dose, dwell time, and treatment time calculations at representative points;
11.12.1.3 The accuracy of isodose plots and graphic displays;
11.12.1.4 The accuracy of the software used to determine radiation source positions from radiographic images; and
11.12.1.5 If the treatment-planning system is different from the treatment-delivery system, the accuracy of electronic transfer of the treatment delivery parameters to the treatment delivery unit from the treatment planning system.
11.12.2 The position indicators in the applicator shall be compared to the actual position of the source or planned dwell positions, as appropriate, at the time of commissioning.
11.12.3 Prior to each patient treatment regimen, the parameters for the treatment shall be evaluated and approved by the authorized user and the Qualified Medical Physicist for correctness through means independent of that used for the determination of the parameters.
11.13 Training.
11.13.1 A registrant shall provide instruction, initially and at least annually, to all individuals who operate the electronic brachytherapy device, as appropriate to the individual's assigned duties, in the operating procedures identified in 11.8. If the interval between patients exceeds one year, retraining of the individuals shall be provided.
11.13.2 In addition to the requirements of 3.3 for therapeutic radiation machine authorized users and 3.4 for Qualified Medical Physicists, these individuals shall also receive device specific instruction initially from the manufacturer, and annually from either the manufacturer or other qualified trainer. The training shall be of a duration recommended by a recognized national professional association with expertise in electronic brachytherapy (when available). In the absence of any training protocol recommended by a national professional association, the manufacturer’s training protocol shall be followed. The training shall include, but nor be limited to:
11.13.2.1 Device-specific radiation safety requirements;
11.13.2.2 Device operation;
11.13.2.3 Clinical use for the types of use approved by the FDA;
11.13.2.4 Emergency procedures, including an emergency drill; and
11.13.2.5 The registrant’s Quality Assurance Program.
11.13.3 A registrant shall retain a record of individuals receiving instruction required by 11.13.1 and 11.13.2 for three (3) years The record shall include a list of the topics covered, the date of the instruction, the name(s) of the attendee(s), and the name(s) of the individual(s) who provided the instruction.
11.14 Mobile Electronic Brachytherapy Service. A registrant providing mobile electronic brachytherapy service shall, as a minimum:
11.14.1 Check all survey instruments before medical use at each address of use or on each day of use, whichever is more restrictive.
11.14.2 Account for the electronic brachytherapy source in the electronic brachytherapy device before departure from the client’s address.
11.14.3 Perform, at each location on each day of use, all of the required quality assurance checks specified 11.11 to assure proper operation of the device.
12.1 A person shall not utilize any device which is designed to electrically generate a source of ionizing radiation to deliver therapeutic radiation dosage, and which is not appropriately regulated under any existing category of therapeutic radiation machine, until:
12.1.1 The applicant or registrant has, at a minimum, provided the Agency with:
12.1.1.1 A detailed description of the device and its intended application(s);
12.1.1.2 Facility design requirements, including shielding and access control;
12.1.1.3 Documentation of appropriate training for authorized user physician(s) and qualified medical physicist(s)
12.1.1.4 Methodology for measurement of dosages to be administered to patients or human research subjects;
12.1.1.5 Documentation regarding calibration, maintenance, and repair of the device, as well as instruments and equipment necessary for radiation safety
12.1.1.6 Radiation safety precautions and instructions; and
12.1.1.7 Other information requested by the Agency in its review of the application; and
12.1.2 The applicant or registrant has received written approval from the Agency to utilize the device in accordance with the regulations and specific conditions the Agency considers necessary for the medical use of the device.
APPENDIX A
INFORMATION ON RADIATION SHIELDING REQUIRED FOR PLAN REVIEWS
1.1 Basic facility information including: name, telephone number and Agency registration number of the individual responsible for preparation of the shielding plan; name and telephone number of the facility supervisor; and the street address [including room number] of the therapeutic radiation machine facility. The plan should also indicate whether this is a new structure or a modification to existing structure(s).
1.2 All wall, floor, and ceiling areas struck by the useful beam shall have primary barriers.
1.3 Secondary barriers shall be provided in all wall, floor, and ceiling areas not having primary barriers.
2.1 In addition to the requirements listed in Section I above, therapeutic radiation machine facilities which produce only photons with a maximum energy less than or equal to 150 kV shall submit shielding plans which contain, as a minimum, the following additional information:
2.2 Equipment specifications, including the manufacturer and model number of the therapeutic radiation machine, as well as the maximum technique factors;
2.3 Maximum design workload for the facility including total weekly radiation output, [expressed in gray (rad) or air kerma at 1 meter], total beam‑on time per day or week, the average treatment time per patient, along with the anticipated number of patients to be treated per day or week;
2.4 A facility blueprint/drawing indicating: scale [0.25 inch = 1 foot is typical]; direction of North; normal location of the therapeutic radiation machine's radiation port(s); the port's travel and traverse limits; general direction(s) of the useful beam; locations of any windows and doors; and the location of the therapeutic radiation machine control panel. If the control panel is located inside the therapeutic radiation machine treatment room, the location of the operator's booth shall be noted on the plan and the operator's station at the control panel shall be behind a protective barrier sufficient to ensure compliance with Part D.1201 of these regulations;
2.5 The structural composition and thickness or lead/concrete equivalent of all walls, doors, partitions, floor, and ceiling of the room(s) concerned;
2.6 The type of occupancy of all adjacent areas inclusive of space above and below the room(s) concerned. If there is an exterior wall, show distance to the closest area(s) where it is likely that individuals may be present; and
2.7 At least one example calculation which shows the methodology used to determine the amount of shielding required for each physical condition [i.e.: primary and secondary/leakage barriers, restricted and unrestricted areas, entry door(s)] and shielding material in the facility:
2.7.1 If commercial software is used to generate shielding requirements, please also identify the software used and the version/ revision date.
2.7.2 If the software used to generate shielding requirements is not in the open literature, please also submit quality control sample calculations to verify the result obtained with the software.
3.1 In addition to the requirements listed in Section I above, therapeutic radiation machine facilities that produce photons with a maximum energy in excess of 150 kV and/or electrons shall submit shielding plans which contain, as a minimum, the following additional information:
3.2 Equipment specifications including the manufacturer and model number of the therapeutic radiation machine, and gray (rad) at the isocenter and the energy(s) and type(s) of radiation produced [i.e.: photon, electron]. The target to isocenter distance shall be specified;
3.3 Maximum design workload for the facility including total weekly radiation output [expressed in gray (rad) at 1 meter], total beam‑on time per day or week, the average treatment time per patient, along with the anticipated number of patients to be treated per day or week;
3.4 Facility blueprint/drawing [including both floor plan and elevation views] indicating relative orientation of the therapeutic radiation machine, scale [0.25 inch = 1 foot is typical], type(s), thickness and minimum density of shielding material(s), direction of North, the locations and size of all penetrations through each shielding barrier [ceiling, walls and floor], as well as details of the door(s) and maze;
3.5 The structural composition and thickness or concrete equivalent of all walls, doors, partitions, floor, and ceiling of the room(s) concerned;
3.6 The type of occupancy of all adjacent areas inclusive of space above and below the room(s) concerned. If there is an exterior wall, show distance to the closest area(s) where it is likely that individuals may be present;
3.7 Description of all assumptions that were in shielding calculations including, but not limited to, design energy [i.e.: room may be designed for 6 MV unit although only a 4 MV unit is currently proposed], work‑load, presence of integral beam‑stop in unit, occupancy and use(s) of adjacent areas, fraction of time that useful beam will intercept each permanent barrier [walls, floor and ceiling] and "allowed" radiation exposure in both restricted and unrestricted areas; and
3.8 At least one example calculation which shows the methodology used to determine the amount of shielding required for each physical condition [i.e.: primary and secondary/leakage barriers, restricted and unrestricted areas, small angle scatter, entry door(s) and maze] and shielding material in the facility:
3.8.1 If commercial software is used to generate shielding requirements, also identify the software used and the version/ revision date; and
3.8.2 If the software used to generate shielding requirements is not in the open literature, also submit quality control sample calculations to verify the result obtained with the software.
4.1 In addition to the requirements listed in Section III above, therapeutic radiation machine facilities that are capable of operating above 10 MV shall submit shielding plans which contain, as a minimum, the following additional information:
4.2 The structural composition, thickness, minimum density and location of all neutron shielding material;
4.3 Description of all assumptions that were used in neutron shielding calculations including, but not limited to, neutron spectra as a function of energy, neutron fluence rate, absorbed dose and dose equivalent (due to neutrons) in both restricted and unrestricted areas;
4.4 At least one example calculation which shows the methodology used to determine the amount of neutron shielding required for each physical condition [i.e.: restricted and unrestricted areas, entry door(s) and maze] and neutron shielding material utilized in the facility:
4.4.1 If commercial software is used to generate shielding requirements, also identify the software used and the version/ revision date; and
4.4.2 If the software used to generate shielding requirements is not in the open literature, also submit quality control sample calculations to verify the result obtained with the software.
4.5 The method(s) and instrumentation that will be used to verify the adequacy of all neutron shielding installed in the facility.
5.1 NCRP Report 49, "Structural Shielding Design and Evaluation for Medical Use of X Rays and Gamma Rays of Energies Up to 10 MeV" (1976).
5.2 NCRP Report 79, "Neutron Contamination from Medical Electron Accelerators" (1984).
5.3 NCRP Report 144, "Radiation Protection for Particle Accelerator Facilities" (2003).
5.4 NCRP Report 151, “Structural Shielding Design and Evaluation for Megavoltage X- and Gamma-Ray Radiotherapy Facilities. (2006).

