


 Authenticated PDF Version
Authenticated PDF Version1124 Control of Organic Compound Emissions
1.0 Test methods.
The owner or operator of any volatile organic compound (VOC) source required to comply with 10.0 through 50.0 of this regulation shall, at the owner's or operator's expense, demonstrate compliance by using the methods of Appendix A through Appendix G of this regulation or alternative methods that are approved by the Department as part of a State Implementation Plan (SIP) or Federal Implementation Plan (FIP) revision and shall meet all the requirements of this appendix.
2.0 Preparation of test plan and quality assurance (QA) program.
At least 30 days before the initiation of a required test under Appendix D of this regulation, the owner or operator shall submit a test plan that shall be approved by the Department before the results of the test are considered acceptable. This test plan shall include the following minimum information:
2.1 The purpose of the proposed test and the applicable provisions of 13.0 through 43.0 of this regulation.
2.2 A detailed description of the facility to be tested, including a line diagram of the facility, locations of test sites, and facility operation conditions for the test.
2.3 A detailed description of the test methods and procedures, equipment, and sampling sites, i.e., a test plan which includes a detailed description of the process and control device operating parameters to be collected during the test.
2.4 A timetable for the following:
2.4.1 Date for the compliance test.
2.4.2 Date of submittal of preliminary results to the Department (not later than 60 days after sample collection).
2.4.3 Date of submittal of final test report (not later than 90 days after completion of on-site sampling).
2.5 Proposed corrective actions should the test results show noncompliance.
2.6 Internal QA program. The internal QA program shall include, at a minimum, the activities planned by routine operators and analysts to provide an assessment of test data precision. An example of internal QA is the sampling and analysis of replicable samples.
2.7 External QA program.
2.7.1 The external QA program shall include, at a minimum, application of plans for a test method performance audit (PA) during the compliance test.
2.7.2 The external QA program may also include systems audits, which include the opportunity for on-site evaluation by the Department of instrument calibration, data validation, sample logging, and documentation of quality control data and field maintenance activities.
2.7.3 The PA's shall consist of blind audit samples provided by the Department and analyzed during the compliance test to provide a measure of test data bias.
2.7.3.1 The Department shall require the owner or operator to analyze QA samples during each compliance test when audit samples are available.
2.7.3.2 Information concerning the availability of audit materials for a specific compliance test may be obtained by contacting the Department.
2.7.3.3 The evaluation criteria applied to the interpretation of the PA results and the subsequent remedial actions required of the owner or operator are the sole responsibility of the Department.
3.0 Process operation.
3.1 Sampling ports, pipes, lines, or appurtenances for collecting samples and data required by the test methods and procedures.
3.2 Safe access to the sample and data collection locations.
3.3 Light, electricity, and the utilities required for sample and data collection.
4.0 Summary of results.
No later than 60 days after the sample collection, the owner or operator shall submit preliminary results to the Department.
5.0 Final report.
No later than 90 days after completion of the on-site sampling, the owner or operator shall submit a test report to the Department. The test report shall include the following minimum information:
5.1 Process description.
5.2 Air pollution capture system and control device description.
5.3 Process conditions during testing, to include operating data for the air pollution control devices (APCD).
5.4 Test results and example calculations.
5.5 Description of sampling locations and test methods.
5.6 QA measures.
5.7 Field and analytical data.
1.0 Sampling procedures shall follow the guidelines presented in "Standard Procedure for Collection of Coating and Ink Samples for VOC Content Analysis by Reference Method 24 and Reference Method 24A," EPA-340/1-91-010.
2.0 The analytical methods and procedures specified below shall be used to determine the VOC content of each coating, as applied:
2.1.1 Method 24 of 40 CFR Part 60, Appendix A (July 1, 1992), shall be used to determine total volatile content, water content, and density of coatings. For determining total volatile content, all samples shall be oven-dried at 110° C for one hour.
2.1.2 To determine the total volatile content, water content, and density of multi-component coatings, the following procedures shall be used in addition to Method 24 of 40 CFR Part 60, Appendix A (July 1, 1992):
2.1.2.1 The components shall be mixed in a storage container in the same proportions as those in the coating, as applied. The mixing shall be accomplished by weighing the components in the proper proportion into a container that is closed between additions and during mixing. About 100 milliliters (ml) of coating shall be prepared in a container just large enough to hold the mixture prior to withdrawing a sample.
2.1.2.2 For determining volatile content, a sample shall be withdrawn from the mixed coating and then transferred to a dish where the sample shall stand for at least one hour, but no more than 24 hours, prior to being oven-dried at 110° C for one hour.
2.1.2.3 For determining the water content and density of multi-component coatings, samples shall be taken from the same 100-ml mixture of coating and shall be analyzed by the appropriate ASTM methods referenced in Method 24 of 40 CFR, Part 60, Appendix A (July 1, 1992).
2.2 Method 24 of 40 CFR Part 60, Appendix A (July 1, 1992), shall be used in determining total volatile content, water content, and density of any flexographic or packaging rotogravure printing ink and related coatings. Alternatively, Method 24A of 40 CFR Part 60, Appendix A (July 1, 1992), may be used.
2.3 Method 24A of 40 CFR Part 60, Appendix A (July 1, 1992), shall be used in determining total volatile content, water content, and density of any publication rotogravure printing ink and related coatings.
2.4 The following additional procedure shall be used in analyzing a coating sample: "Standard Procedure for Analysis of Coating and Ink Samples," EPA-340/1-91-011.
3.0 Use of adaptations to test methods.
Use of an adaptation to any of the analytical methods specified in 2.0 of this appendix shall be approved by the Department on a case-by-case basis. An owner or operator shall submit sufficient documentation for the Department to find that the analytical methods specified in 2.1, 2.2, and 2.3 of this appendix will yield inaccurate results and that the proposed adaptation is appropriate.
4.0 Each sample collected for analysis shall meet the following criteria:
4.1 Each sample shall be at least 250 ml (eight fluid ounces [oz]) taken into a 250-ml (eight-oz) container at a location and time such that the sample will be representative of the coating or ink, as applied (i.e., the sample shall include any dilution solvent or VOC added during the manufacturing process).
4.2 If a sample larger than 250 ml (eight oz) is obtained, the sample container shall be of a size such that the sample completely fills the container.
4.3 The container shall be tightly sealed immediately after the sample is taken.
4.4 Any solvent or other VOC added after the sample is taken shall be measured and accounted for in the calculations in 3.0 of this appendix.
4.5 For multi-component coatings, separate samples of each component shall be obtained.
5.0 Calculations for determining the VOC content of coatings and inks from data as determined by Method 24 or 24A of 40 CFR Part 60, Appendix A (July 1, 1992), shall follow the guidance provided in the following documents:
5.1 "A Guideline for Surface Coating Calculations," EPA-340/1-86-016.
5.2 "Procedures for Certifying Quantity of Volatile Organic Compounds Emitted by Paint, Ink and Other Coatings," (Revised June 1986) EPA-450/3-84-019.
1.0 Daily-weighted average.
The daily-weighted average VOC content, in units of mass of VOC per unit volume of coating, excluding water and exempt compounds, as applied, of the coatings used on a day on a coating unit, line, or operation shall be calculated using the following equation:
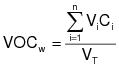 (C-1)
(C-1)VOCw = The daily-weighted average VOC content of the coatings, as applied, used on a coating unit, line, or operation in units of kilograms of VOC per liter of coating (kg VOC/L) (pounds of VOC per gallon of coating [lb VOC/gal]), excluding water and exempt compounds.
Vi = The volume of each coating, as applied, each day on a coating unit, line, or operation in units of L (gal), excluding water and exempt compounds.
Ci = The VOC content of each coating, as applied, each day on a coating unit, line, or operation in units of kg VOC/L of coating (lb VOC/gal), excluding water and exempt compounds.
VT = The total volume of all coating, as applied, each day on a coating unit, line, or operation in units of L (gal), excluding water and exempt compounds.
2.0 [Reserved]
3.0 Overall emission reduction efficiency for control systems.
The overall emission reduction efficiency needed to demonstrate compliance is determined each day as follows:
3.1 Obtain the emission limitation from the applicable provisions in 10.0 through 50.0 of this regulation.
3.2 Calculate the emission limitation on a solids basis according to the following equation:
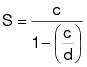 (C-2)
(C-2)c = The VOC emission limitation in terms of kg VOC/L of coating (lb/gal), excluding water and exempt compounds.
d = The density of VOC for converting emission limitation to a solids basis. The density equals 0.882 kg/L (7.36 lb/gal).
3.3 Calculate the required overall emission reduction efficiency of the control system for the day according to the following equation:
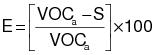 (C-3)
(C-3) VOCa = (1) The maximum VOC content of the coatings, as applied, used each day on the subject coating unit, line, or operation, in units of kg VOC/L of coating solids (lb VOC/gal), as determined by the applicable test methods and procedures specified in Appendix B of this regulation. (2) Alternatively, the daily-weighted average VOC content, as applied, of the coatings used each day on the subject coating unit, line, or operation, in units of kg VOC/L of coating solids (lb VOC/gal), as determined by the applicable test methods and procedures specified in Appendix B of this regulation and the procedure in 3.4 of this appendix.
3.4 The daily-weighted average VOC content, as applied, of the coatings used on a coating unit, line, or operation in units of mass of VOC per unit volume of coating solids shall be calculated by the following equation:
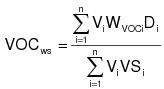 (C-4)
(C-4)VOCws = The daily-weighted average VOC content, as applied, of the coatings used on a coating unit, line, or operation in units of mass of VOC per unit volume of coating solids.
Vi = The volume of each coating (i), as applied, used in a day on a coating unit, line, or operation in units of liters (L) (gallons [gal]).
WVOCi = The weight fraction of VOC in each coating (i), as applied, used in a day on a coating unit, line, or operation in units of kg VOC/kg coating (lb/lb).
Di = The density of each coating (i) as applied, used in a day on a coating unit, line, or operation in units of kg coating/L of coating (lb/gal).
VSi = The volume fraction solids content of each coating (i), as applied, used in a day on a coating unit, line, or operation in units of L solids/L coating (gal/gal).
1.0 Determining the efficiency of volatile organic compound (VOC) capture systems.
1.1 For purposes of this appendix, the following definitions and abbreviations apply:
“BE” is a building or room enclosure that contains a process that emits VOCs. If a BE is to substitute for a PTE or TTE, the appropriate requirements given in this appendix shall be met.
“Gas/gas method” means either of two methods for determining capture that rely only on gas phase measurements. One method requires construction of a temporary total enclosure (TTE) to ensure all potential fugitive emissions are measured while the other method uses the room or building that houses the source as an enclosure.
“Hood” means a partial enclosure or canopy for capturing and exhausting, by means of a draft, the organic vapors or other fumes rising from a coating process or other source.
“Liquid/gas Method” means either of two methods for determining capture that require both gas phase and liquid phase measurements and analysis. One liquid/gas method requires construction of a temporary enclosure, and the other uses the building or room that houses the facility as an enclosure.
“PTE” is a permanent total enclosure, which contains a process that emits VOC and meets the specifications given in this appendix, Method 30.
“TTE” is a temporary total enclosure that is built around a process that emits VOC and meets the specifications given in this appendix, Method 30.
1.2 Applicability.
1.2.1 The requirements of 1.3 of this appendix shall apply to all regulated VOC emitting processes using a control system except as provided below.
1.2.2 If a source owner or operator installs a PTE that meets EPA specifications, and that directs all VOC to a control device, the capture efficiency is assumed to be 100%, and the source is exempted from the requirements described in 1.3 of this appendix. The method given in this appendix shall be used to determine whether a structure is a PTE. This does not exempt a source from performing any control device efficiency testing required under this regulation. In addition, source shall demonstrate that all criteria for a PTE are met during the testing for capture efficiency.
1.2.3 If a source owner or operator uses a control device designed to collect and recover VOC (e.g., carbon adsorber), an explicit measurement of capture efficiency is not necessary if the conditions given below are met. The overall emission reduction efficiency of the control system shall be determined each day by directly comparing the input liquid VOC (L) to the recovered liquid VOC. The procedure for use in this situation is specified in 40 CFR 60.433 (July 1, 1992) with the following modifications:
1.2.3.1 The source owner or operator shall obtain data each day for the solvent usage and solvent recovery and determine the solvent recovery efficiency of the system each day using a seven-day rolling period. The recovery efficiency for each day is computed as the ratio of the total recovered solvent for that day and the prior six consecutive operating days to the total solvent usage for the same seven-day period used for the recovered solvent, rather than a 30-day weighted average as given in 40 CFR 60.433 (July 1, 1992). This ratio shall be expressed as a percentage. This shall be done within 72 hours following each 24-hour period. A source that believes that the seven -day rolling period is not appropriate may use the method provided in Appendix I of this regulation to determine an alternative multiday rolling period. In no event shall the rolling period determined under this method exceed a 30-day rolling period.
1.2.3.2 If the solvent recovery system controls multiple process lines, the source owner or operator shall demonstrate that the overall control (i.e., the total recovered solvent VOC divided by the sum of liquid VOC input to all process lines venting to the control system) meets or exceeds the most stringent standard applicable for any process line venting to the control system.
1.3 Specific Requirements.
1.3.1 The capture efficiency shall be measured using one of the four protocols given in 1.3.3.1 through 1.3.3.5 of this appendix.
1.3.2 Any error margin associated with a test protocol may not be incorporated into the results of a capture efficiency test.
1.3.3 Any source required to comply with this appendix shall use one of the following protocols to measure capture efficiency, unless a suitable alternative protocol is approved by the U.S. EPA as part of a SIP or FIP revision:
1.3.3.1 Gas/gas method using TTE. Method 30, given in this appendix, shall be used to determine whether a temporary enclosure is a TTE. The capture efficiency equation to be used for this protocol is:
Either Method 30B or Method 30C of this appendix is used to obtain G. Method 30D of this appendix is used to obtain F.
1.3.3.2 Liquid/gas method using TTE. Method 30, given in this appendix, shall be used to determine whether a temporary enclosure is a TTE. The capture efficiency equation to be used for this protocol is:
1.3.3.3 Gas/gas method using the building or room (BE) in which the source is located as the enclosure and in which G and F are measured while operating only the source to be tested. All fans and blowers in the building or room shall be operated as they would be under normal production. The capture efficiency equation to be used for this protocol is:
Either Method 30B or Method 30C of this appendix is used to obtain G. Method 30E of this appendix is used to obtain FB .
1.3.3.4 Liquid/gas method using the building or room (BE) in which the source is located as the enclosure and in which L and F are measured while operating only the source to be tested. All fans and blowers in the building or room shall be operated as they would under normal production. The capture efficiency equation to be used for this protocol is:
1.3.3.5 Liquid/gas method using the collection device to determine the mass of gas collected. The Capture Efficiency equation to be used for this process is:
1.4 Recordkeeping and Reporting.
1.4.1 All sources complying with this appendix shall maintain on file a copy of the capture efficiency protocol submitted to the Department. All results of appropriate test methods and CE protocols shall be reported to the Department within 60 days of the test date. A copy of the results shall be kept on file with the source.
1.4.2 If any changes are made to capture or control equipment, the source is required to notify the Department within 30 days of these changes and a new capture efficiency or control device destruction or removal efficiency test may be required.
2.0 Determining the destruction or removal efficiency of incinerators and carbon adsorbers.
2.1 Testing.
2.1.1 The control device destruction or removal efficiency shall be determined from data obtained by simultaneously measuring the inlet and outlet gas-phase VOC concentrations and gas volumetric flow rates in accordance with the gas-phase test methods specified in Appendix E of this regulation. The control device destruction or removal efficiency shall be calculated using the following equation:
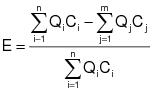 (D-6)
(D-6)Qi = Volumetric flow rate of the effluent gas flowing through stack i entering the control device, dry standard cubic meters per hour (dscm/hr).
Ci = Concentration of VOC (as carbon) in the effluent gas flowing through stack i entering the control device, parts per million by volume (ppmv).
Qj = Volumetric flow rate of the effluent gas flowing through stack j leaving the control device, dscmh.
Cj = Concentration of VOC (as carbon) in the effluent gas flowing through stack j leaving the control device, ppmv.
2.1.2 A source using a PTE (or a BE as a PTE) shall demonstrate that this enclosure meets the requirements given in Method 30 of this appendix for a PTE during any testing of a control device.
2.1.3 A source using a TTE (or a BE as a TTE) shall demonstrate that this enclosure meets the requirements given in Method 30 of this appendix for a TTE during testing of a control device. The source shall also provide documentation that the quality assurance criteria for a TTE have been achieved.
2.2 Monitoring.
2.2.1 Any owner or operator who uses an incinerator or regenerative carbon adsorber to comply with any part of this regulation shall install, calibrate, certify to the Department, operate, and maintain continuous monitoring equipment. The continuous monitoring equipment shall monitor the following parameters:
2.2.1.1 Combustion chamber temperature of each thermal incinerator or afterburner.
2.2.1.2 Temperature rise immediately before the catalyst bed and across each catalytic incinerator bed.
2.2.1.3 The VOC concentration of the outlet from each carbon adsorption bed.
2.2.2 The continuous temperature monitoring equipment must be equipped with a continuous recorder and have an accuracy of ±1% of the combustion temperature being measured expressed in degrees Celsius (°C) or ±0.5°C, whichever is greater.
2.2.3 The owner or operator shall ensure that the quality assurance measures in 10.0 of Appendix G of this regulation and the quality control procedures in Appendix H of this regulation are met.
3.0 Determining the overall emission reduction efficiency.
The overall emission reduction efficiency of the emission control system shall be determined each day as the product of the capture efficiency, as determined using the capture efficiency test method in this appendix, and the control device destruction or removal efficiency; or for each solvent recovery system, by the test protocol described in 1.2.3.1 of this appendix for comparing liquid input to liquid VOC recovery. The results of the capture efficiency test and control device destruction or removal efficiency test remain valid for each day until a subsequent test is performed. The results of any valid test may be used for each day until superseded by the results of a valid test subsequently performed.
1.0 Introduction.
1.1 Applicability. This procedure is used to determine whether a permanent or temporary enclosure meets the criteria for a total enclosure.
1.2 Principle. An enclosure is evaluated against a set of criteria. If the criteria are met and if all the exhaust gases from the enclosure are ducted to a control device, then the VOC CE is assumed to be 100%, and CE need not be measured. However, if part of the exhaust gas stream is not ducted to a control device, CE must be determined.
1.3 Note. An evaluation of the proposed building materials is recommended to minimize any potential hazards.
2.0 Definitions.
2.1 Natural Draft Opening (NDO). Any permanent opening in the enclosure that remains open during operation of the facility and is not connected to a duct in which a fan is installed.
2.2 Permanent Total Enclosure (PTE). A permanently installed enclosure that completely surrounds a source of emissions such that all VOC emissions are captured and contained for discharge to a control device.
2.3 Temporary Total Enclosure (TTE). A temporarily installed enclosure that completely surrounds a source of emissions such that all fugitive VOC emissions are captured and contained for discharge through ducts that allow for the accurate measurement of fugitive VOC emissions.
3.0 Criteria for Temporary Total Enclosure
3.1 Any NDO shall be at least four equivalent opening diameters from each VOC emitting point unless otherwise specified by the Department.
3.2 Any exhaust point from the enclosure shall be at least four equivalent duct or hood diameters from each NDO.
3.3 The total area of all NDO's shall not exceed 5% of the surface area of the enclosure's four walls, floor, and ceiling.
3.4 The average facial velocity (FV) of air through all NDO's shall be at least 3,600 m/hr (200 fpm). The direction of air flow through all NDO's shall be into the enclosure.
3.5 All access doors and windows whose areas are not included in 3.3 of this method and are not included in the calculation in 3.4 of this method shall be closed during routine operation of the process.
4.0 Criteria for a Permanent Total Enclosure
4.1 Same as 3.1 and 3.3 through 3.5 of this method.
4.2 All VOC emissions must be captured and contained for discharge through a control device.
5.0 Procedure.
5.1 Determine the equivalent diameters of the NDO's and determine the distances from each VOC emitting point to all NDO's. Determine the equivalent diameter of each exhaust duct or hood and its distance to all NDO's. Calculate the distances in terms of equivalent diameters. The number of equivalent diameters shall be at least four.
5.2 Measure the total area (AT) of the enclosure and the total area (AN) of all NDO's in the enclosure. Calculate the NDO to enclosure area ratio (NEAR) as follows:
The NEAR must be <0.05.
5.3 Measure the volumetric flow rate, corrected to standard conditions, of each gas stream exiting the enclosure through an exhaust duct or hood using EPA Method 2. In some cases (e.g., when the building is the enclosure), it may be necessary to measure the volumetric flow rate, corrected to standard conditions, of each gas stream entering the enclosure through a forced makeup air duct using Method 2. Calculate FV using the following equation:
QO = the sum of the volumetric flow from all gas streams exiting the enclosure through an exhaust duct or hood.
QI = the sum of the volumetric flow from all gas streams into the enclosure through a forced makeup air duct; zero, if there is no forced makeup air into the enclosure.
AN = total area of all NDO's in enclosure.
The FV shall be at least 3,600 m/hr (200 fpm). Alternatively, measure the pressure differential across the enclosure. A pressure drop of 0.0075 mm Hg (0.004 in. H2O) corresponds to an FV of 3,600 m/hr (200 fpm).
5.4 Verify that the direction of air flow through all NDO's is inward. Streamers, smoke tubes, or tracer gases may be used. Strips of plastic wrapping film have also been found to be effective. Monitor the direction of air flow for at least one hour, with checks made no more than 10 minutes apart.
6.0 Quality Assurance.
6.1 The success of this method lies in designing the TTE to simulate the conditions that exist without the TTE (i.e., the effect of the TTE on the normal flow patterns around the affected facility or the amount of fugitive VOC emissions should be minimal). The TTE must enclose the application stations, coating reservoirs, and all areas from the application station to the oven. The oven does not have to be enclosed if it is under negative pressure. The NDO's of the temporary enclosure and a fugitive exhaust fan must be properly sized and placed.
6.2 Estimate the ventilation rate of the TTE that best simulates the conditions that exist without the TTE (i.e., the effect of the TTE on the normal flow patterns around the affected facility or the amount of fugitive VOC emissions should be minimal). Measure the concentration (CG) and flow rate (QG) of the captured gas stream, specify a safe concentration (CF) for the fugitive gas stream, estimate the CE, and then use a plot available from the Department to determine the volumetric flow rate of the fugitive gas stream (QF). A fugitive VOC emission exhaust fan that has a variable flow control is desirable.
6.3 Monitor the concentration of VOC into the capture device without the TTE. To minimize the effect of temporal variation on the captured emissions, the baseline measurement should be made over as long a time period as practical. However, the process conditions must be the same for the measurement in 6.5 of this appendix as they are for this baseline measurement. This may require short measuring times for this quality control check before and after the construction of the TTE.
6.4 After the TTE is constructed, monitor the VOC concentration inside the TTE. This concentration shall not continue to increase, and must not exceed the safe level according to Occupational Safety and Health Administration requirements for permissible exposure limits. An increase in VOC concentration indicates poor TTE design or poor capture efficiency.
6.5 Monitor the concentration of VOC into the capture device with the TTE. To limit the effect of the TTE on the process, the VOC measurement with and without the TTE must be within ±10%. If the measurements do not agree, adjust the ventilation rate from the TTE until they agree within 10%.
1.0 Introduction
1.1 Applicability. This procedure is applicable for determining the input of VOC. It is intended to be used in the development of liquid/gas protocols for determining VOC CE for surface coating and printing operations.
1.2 Principle. The amount of VOC introduced to the process (L) is the sum of the products of the weight (W) of each VOC containing liquid (ink, paint, solvent, etc.) used and its VOC content (V). A sample of each VOC containing liquid is analyzed with a flame ionization analyzer (FIA) to determine V.
1.3 Estimated Measurement Uncertainty. The measurement uncertainties are estimated for each VOC containing liquid as follows: W = ±2.0% and V = ±12.0%. Based on these numbers, the probable uncertainty for L is estimated at about ±12.2% for each VOC containing liquid.
1.4 Sampling Requirements. A CE test shall consist of at least three sampling runs. Each run shall cover at least one complete production cycle, but shall be at least three hours long. The sampling time for each run need not exceed eight hours, even if the production cycle has not been completed. Alternative sampling times may be used with the approval of the Department.
1.5 Notes. Because this procedure is often applied in highly explosive areas, caution and care should be exercised in choosing, installing, and using the appropriate equipment. Mention of trade names or company products does not constitute endorsement. All gas concentrations (percent, ppm) are by volume, unless otherwise noted.
2.0 Apparatus and Reagents
2.1 Liquid Weight.
2.1.1 Balances/Digital Scales. To weigh drums of VOC containing liquids to within 0.2 lb.
2.1.2 Volume Measurement Apparatus (Alternative). Volume meters, flow meters, density measurement equipment, etc., as needed to achieve the same accuracy as direct weight measurements.
2.2 VOC Content (FIA Technique). The following equipment is required:
2.2.1 Sample Collection Can. An appropriately-sized metal can to be used to collect VOC containing materials. The can must be constructed in such a way that it can be grounded to the coating container.
2.2.2 Needle Valves. To control gas flow.
2.2.3 Regulators. For carrier gas and calibration gas cylinders.
2.2.4 Tubing. Teflon or stainless steel tubing with diameters and lengths determined by connection requirements of equipment. The tubing between the sample oven outlet and the FIA shall be heated to maintain a temperature of 120 ± 5°C.
2.2.5 Atmospheric Vent. A tee and 0- to 0.5-liter/min rotameter placed in the sampling line between the carrier gas cylinder and the VOC sample vessel to release the excess carrier gas. A toggle valve placed between the tee and the rotameter facilitates leak tests of the analysis system.
2.2.6 Thermometer. Capable of measuring the temperature of the hot water bath to within 1°C.
2.2.7 Sample Oven. Heated enclosure, containing calibration gas coil heaters, critical orifice, aspirator, and other liquid sample analysis components, capable of maintaining a temperature of 120 ± 5°C.
2.2.8 Gas Coil Heaters. Sufficient lengths of stainless steel or Teflon tubing to allow zero and calibration gases to be heated to the sample oven temperature before entering the critical orifice or aspirator.
2.2.9 Water Bath. Capable of heating and maintaining a sample vessel temperature of 100 ±5°C.
2.2.10 Analytical Balance. To measure ±0.001 g.
2.2.11 Disposable Syringes. 2-cc or 5-cc.
2.2.12 Sample Vessel. Glass, 40-ml septum vial. A separate vessel is needed for each sample.
2.2.13 Rubber Stopper. Two-hole stopper to accommodate 3.2-mm (1/8-in.) Teflon tubing, appropriately sized to fit the opening of the sample vessel. The rubber stopper should be wrapped in Teflon tape to provide a tighter seal and to prevent any reaction of the sample with the rubber stopper. Alternatively, any leak-free closure fabricated of nonreactive materials and accommodating the necessary tubing fittings may be used.
2.2.14 Critical Orifices. Calibrated critical orifices capable of providing constant flow rates from 50 to 250 ml/min at known pressure drops. Sapphire orifice assemblies (available from O'Keefe Controls Company) and glass capillary tubing have been found to be adequate for this application.
2.2.15 Vacuum Gauge. Zero to 760-mm (0 to 30-in.) Hg U-Tube manometer or vacuum gauge.
2.2.16 Pressure Gauge. Bourdon gauge capable of measuring the maximum air pressure at the aspirator inlet (e.g., 100 psig, 690 kilo-Pascals).
2.2.17 Aspirator. A device capable of generating sufficient vacuum at the sample vessel to create critical flow through the calibrated orifice when sufficient air pressure is present at the aspirator inlet. The aspirator must also provide sufficient sample pressure to operate the FIA. The sample is also mixed with the dilution gas within the aspirator.
2.2.18 Soap Bubble Meter. Of an appropriate size to calibrate the critical orifices in the system.
2.2.19 Organic Concentration Analyzer. An FIA with a span value of 1.5 times the expected concentration as propane; however, other span values may be used if it can be demonstrated that they would provide more accurate measurements. The FIA instrument should be the same instrument used in the gaseous analyses adjusted with the same fuel, combustion air, and sample back-pressure (flow rate) settings. The system shall be capable of meeting or exceeding the following specifications:
2.2.19.1 Zero Drift. Less than ±3.0% of the span value.
2.2.19.2 Calibration Drift. Less than ±3.0% of the span value.
2.2.19.3 Calibration Error. Less than ±5.0% of the calibration gas value.
2.2.20 Integrator/Data Acquisition System. An analog or digital device or computerized data acquisition system used to integrate the FIA response or compute the average response and record measurement data. The minimum data sampling frequency for computing average or integrated values is one measurement value every 5 seconds. The device shall be capable of recording average values at least once per minute.
2.2.21 Chart Recorder (Optional). A chart recorder or similar device is recommended to provide a continuous analog display of the measurement results during the liquid sample analysis.
2.2.22 Calibration and Other Gases. Gases used for calibration, fuel, and combustion air (if required) are contained in compressed gas cylinders. All calibration gases shall be traceable to National Institute of Standards and Technology standards and shall be certified by the manufacturer to ±1% of the tag value. Additionally, the manufacturer of the cylinder should provide a recommended shelf life for each calibration gas cylinder over which the concentration does not change more than ±2% from the certified value. For calibration gas values not generally available, alternative methods for preparing calibration gas mixtures, such as dilution systems, may be used with the approval of the Department.
2.2.22.1 Fuel. A 40% H2/60% He or 40% H2/60% N2 gas mixture is recommended to avoid an oxygen synergism effect that reportedly occurs when oxygen concentration varies significantly from a mean value.
2.2.22.2 Carrier Gas. High purity air with less than one ppm of organic material (as propane) or less than 0.1% of the span value, whichever is greater.
2.2.22.3 FIA Linearity Calibration Gases. Low-, mid-and high-range gas mixture standards with nominal propane concentrations of 20-30, 45-55, and 70-80% of the span value in air, respectively. Other calibration values and other span values may be used if it can be shown to the Department's satisfaction that more accurate measurements would be achieved.
2.2.22.4 System Calibration Gas. Gas mixture standard containing propane in air, approximating the undiluted VOC concentration expected for the liquid samples.
3.0 Determination of Liquid Input Weight
3.1 Weight Difference. Determine the amount of material introduced to the process as the weight difference of the feed material before and after each sampling run. In determining the total VOC containing liquid usage, account for:
3.1.1 Identify all points where VOC containing liquids are introduced to the process. To obtain an accurate measurement of VOC containing liquids, start with an empty fountain (if applicable). After completing the run, drain the liquid in the fountain back into the liquid drum (if possible) and weigh the drum again. Weigh the VOC containing liquids to ±0.5% of the total weight (full) or ±0.1% of the total weight of VOC containing liquid used during the sample run, whichever is less. If the residual liquid cannot be returned to the drum, drain the fountain into a pre-weighed empty drum to determine the final weight of the liquid.
3.1.2 If it is not possible to measure a single representative mixture, then weigh the various components separately (e.g., if solvent is added during the sampling run, weigh the solvent before it is added to the mixture). If a fresh drum of VOC containing liquid is needed during the run, then weigh both the empty drum and fresh drum.
3.2 Volume Measurement (Alternative). If direct weight measurements are not feasible, the tester may use volume meters and flow rate meters (and density measurements) to determine the weight of liquids used if it can be demonstrated that the technique produces results equivalent to the direct weight measurements. If a single representative mixture cannot be measured, measure the components separately.
4.0 Determination of VOC Content in Input Liquids
4.1 Collection of Liquid Samples.
4.1.1 Collect a 100-ml or larger sample of the VOC containing liquid mixture at each application location at the beginning and end of each test run. A separate sample should be taken of each VOC containing liquid added to the application mixture during the test run. If a fresh drum is needed during the sampling run, then obtain a sample from the fresh drum.
4.1.2 When collecting the sample, ground the sample container to the coating drum. Fill the sample container as close to the rim as possible to minimize the amount of headspace.
4.1.3 After the sample is collected, seal the container so the sample cannot leak out or evaporate.
4.1.4 Label the container to clearly identify the contents.
4.2 Liquid Sample VOC Content.
4.2.1 Assemble the liquid VOC content analysis system.
4.2.2 Permanently identify all of the critical orifices that may be used. Calibrate each critical orifice under the expected operating conditions (i.e., sample vacuum and temperature) against a volume meter as described in 5.3 of this method.
4.2.3 Label and tare the sample vessels (including the stoppers and caps) and the syringes.
4.2.4 Install an empty sample vessel and perform a leak test of the system. Close the carrier gas valve and atmospheric vent and evacuate the sample vessel to 250 mm (10 in.) Hg absolute or less using the aspirator. Close the toggle valve at the inlet to the aspirator and observe the vacuum for at least one minute. If there is any change in the sample pressure, release the vacuum, adjust or repair the apparatus as necessary, and repeat the leak test.
4.2.5 Perform the analyzer calibration and linearity checks according to the procedure in 5.1 of this method. Record the responses to each of the calibration gases and the back-pressure setting of the FIA.
4.2.6 Establish the appropriate dilution ratio by adjusting the aspirator air supply or substituting critical orifices. Operate the aspirator at a vacuum of at least 25 mm (one inch) Hg greater than the vacuum necessary to achieve critical flow. Select the dilution ratio so that the maximum response of the FIA to the sample does not exceed the high-range calibration gas.
4.2.7 Perform system calibration checks at two levels by introducing compressed gases at the inlet to the sample vessel while the aspirator and dilution devices are operating. Perform these checks using the carrier gas (zero concentration) and the system calibration gas. If the response to the carrier gas exceeds ±0.5% of span, clean or repair the apparatus and repeat the check. Adjust the dilution ratio as necessary to achieve the correct response to the upscale check, but do not adjust the analyzer calibration. Record the identification of the orifice, aspirator air supply pressure, FIA back-pressure, and the responses of the FIA to the carrier and system calibration gases.
4.2.8 After completing the above checks, inject the system calibration gas for approximately 10 minutes. Time the exact duration of the gas injection using a stopwatch. Determine the area under the FIA response curve and calculate the system response factor based on the sample gas flow rate, gas concentration, and the duration of the injection as compared to the integrated response using Equations D-30A-2 and D-30A-3 of this method.
4.2.9 Verify that the sample oven and sample line temperatures are 120 ±5°C and that the water bath temperature is 100 ± 5°C.
4.2.10 Fill a tared syringe with approximately one g of the VOC containing liquid and weigh it. Transfer the liquid to a tared sample vessel. Plug the sample vessel to minimize sample loss. Weigh the sample vessel containing the liquid to determine the amount of sample actually received. Also, as a quality control check, weigh the empty syringe to determine the amount of material delivered. The two coating sample weights should agree within 0.02 g. If not, repeat the procedure until an acceptable sample is obtained.
4.2.11 Connect the vessel to the analysis system. Adjust the aspirator supply pressure to the correct value. Open the valve on the carrier gas supply to the sample vessel and adjust it to provide a slight excess flow to the atmospheric vent. As soon as the initial response of the FIA begins to decrease, immerse the sample vessel in the water bath. (Applying heat to the sample vessel too soon may cause the FIA response to exceed the calibrated range of the instrument and, thus, invalidate the analysis.)
4.2.12 Continuously measure and record the response of the FIA until all of the volatile material has been evaporated from the sample and the instrument response has returned to the baseline (i.e., response less than 0.5% of the span value). Observe the aspirator supply pressure, FIA back-pressure, atmospheric vent, and other system operating parameters during the run; repeat the analysis procedure if any of these parameters deviate from the values established during the system calibration checks in 4.2.7 of this method. After each sample, perform the drift check described in 5.2 of this method. If the drift check results are acceptable, calculate the VOC content of the sample using the equations in 7.0 of this method. Integrate the area under the FIA response curve, or determine the average concentration response and the duration of sample analysis.
5.0 Calibration and Quality Assurance
5.1 FIA Calibration and Linearity Check. Make necessary adjustments to the air and fuel supplies for the FIA and ignite the burner. Allow the FIA to warm up for the period recommended by the manufacturer. Inject a calibration gas into the measurement system and adjust the back-pressure regulator to the value required to achieve the flow rates specified by the manufacturer. Inject the zero- and the high-range calibration gases and adjust the analyzer calibration to provide the proper responses. Inject the low- and mid-range gases and record the responses of the measurement system. The calibration and linearity of the system are acceptable if the responses for all four gases are within 5% of the respective gas values. If the performance of the system is not acceptable, repair or adjust the system and repeat the linearity check. Conduct a calibration and linearity check after assembling the analysis system and after a major change is made to the system.
5.2 Systems Drift Checks. After each sample, repeat the system calibration checks in 4.2.7 of this method before any adjustments to the FIA or measurement system are made. If the zero or calibration drift exceeds ±3% of the span value, discard the result and repeat the analysis.
5.3 Critical Orifice Calibration.
5.3.1 Each critical orifice must be calibrated at the specific operating conditions under which it will be used. Therefore, assemble all components of the liquid sample analysis system as they are to be used. A stopwatch is also required.
5.3.2 Turn on the sample oven, sample line, and water bath heaters, and allow the system to reach the proper operating temperature. Adjust the aspirator to a vacuum of 380 mm (15 in.) Hg vacuum. Measure the time required for one soap bubble to move a known distance and record barometric pressure.
5.3.3 Repeat the calibration procedure at a vacuum of 406 mm (16 in.) Hg and at 25-mm (1-in.) Hg intervals until three consecutive determinations provide the same flow rate. Calculate the critical flow rate for the orifice in ml/min at standard conditions. Record the vacuum necessary to achieve critical flow.
6.0 Nomenclature
AL = area under the response curve of the liquid sample, area count.
AS = area under the response curve of the calibration gas, area count.
CS = actual concentration of system calibration gas, ppm propane.
K = 1.830x10-9 g/(ml-ppm).
ML = mass of liquid sample delivered to the sample vessel, g.
0S = total gas injection time for system calibration gas during integrator calibration, min.
VFj = final VOC fraction of VOC containing liquid j.
VIj = initial VOC fraction of VOC containing liquid j.
VAj = VOC fraction of VOC containing liquid j added during the run.
WFj = weight of VOC containing liquid j remaining at end of the run, kg.
WIj = weight of VOC containing liquid j at beginning of the run, kg.
WAj = weight of VOC containing liquid j added during the run, kg.
7.0 Calculations
7.1 Total VOC Content of the Input VOC Containing Liquid.
7.2 Liquid Sample Analysis System Response Factor for Systems Using Integrators, Grams/Area Count.
7.3 VOC Content of the Liquid Sample.
1.0 Introduction
1.1 Applicability. This procedure is applicable for determining the VOC content of captured gas streams. It is intended to be used in the development of liquid/gas or gas/gas protocols for determining VOC CE for surface coating and printing operations. The procedure may not be acceptable in certain site-specific situations [e.g., when: (1) direct-fired heaters or other circumstances affect the quantity of VOC at the control device inlet; and (2) particulate organic aerosols are formed in the process and are present in the captured emissions].
1.2 Principle. The amount of VOC captured (G) is calculated as the sum of the products of the VOC content (CGj), the flow rate (QGj), and the sample time (0C) from each captured emissions point.
1.3 Estimated Measurement Uncertainty. The measurement uncertainties are estimated for each captured or fugitive emissions point as follows: QGj = ±5.5% and CGj = ±5.0%. Based on these numbers, the probable uncertainty for G is estimated at about ±7.4%.
1.4 Sampling Requirements. A CE test shall consist of at least three sampling runs. Each run shall cover at least one complete production cycle, but shall be at least three hours long. The sampling time for each run need not exceed eight hours, even if the production cycle has not been completed. Alternative sampling times may be used with the approval of the Department.
1.5 Notes. Because this procedure is often applied in highly explosive areas, caution and care should be exercised in choosing, installing, and using the appropriate equipment. Mention of trade names or company products does not constitute endorsement. All gas concentrations (percent, ppm) are by volume, unless otherwise noted.
2.0 Apparatus and Reagents
2.1 Gas VOC Concentration. The main components are as follows:
2.1.1 Sample Probe. Stainless steel or equivalent. The probe shall be heated to prevent VOC condensation.
2.1.2 Calibration Valve Assembly. Three-way valve assembly at the outlet of the sample probe to direct the zero and calibration gases to the analyzer. Other methods, such as quick-connect lines, to route calibration gases to the outlet of the sample probe are acceptable.
2.1.3 Sample Line. Stainless steel or Teflon tubing to transport the sample gas to the analyzer. The sample line must be heated to prevent condensation.
2.1.4 Sample Pump. A leak-free pump, to pull the sample gas through the system at a flow rate sufficient to minimize the response time of the measurement system. The components of the pump that contact the gas stream shall be constructed of stainless steel or Teflon. The sample pump must be heated to prevent condensation.
2.1.5 Sample Flow Rate Control. A sample flow rate control valve and rotameter, or equivalent, to maintain a constant sampling rate within 10 %. The flow rate control valve and rotameter must be heated to prevent condensation. A control valve may also be located on the sample pump bypass loop to assist in controlling the sample pressure and flow rate.
2.1.6 Sample Gas Manifold. Capable of diverting a portion of the sample gas stream to the FIA, and the remainder to the bypass discharge vent. The manifold components shall be constructed of stainless steel or Teflon. If captured or fugitive emissions are to be measured at multiple locations, the measurement system shall be designed to use separate sampling probes, lines, and pumps for each measurement location and a common sample gas manifold and FIA. The sample gas manifold and connecting lines to the FIA must be heated to prevent condensation.
2.1.7 Organic Concentration Analyzer. An FIA with a span value of 1.5 times the expected concentration as propane; however, other span values may be used if it can be demonstrated to the Department's satisfaction that they would provide more accurate measurements. The system shall be capable of meeting or exceeding the following specifications:
2.1.7.1 Zero Drift. Less than ±3.0% of the span value.
2.1.7.2 Calibration Drift. Less than ±3.0% of the span value.
2.1.7.3 Calibration Error. Less than ±5.0% of the calibration gas value.
2.1.7.4 Response Time. Less than 30 seconds.
2.1.8 Integrator/Data Acquisition System. An analog or digital device, or computerized data acquisition system used to integrate the FIA response or compute the average response and record measurement data. The minimum data sampling frequency for computing average or integrated values is one measurement value every five seconds. The device shall be capable of recording average values at least once per minute.
2.1.9 Calibration and Other Gases. Gases used for calibration, fuel, and combustion air (if required) are contained in compressed gas cylinders. All calibration gases shall be traceable to National Institute of Standards and Technology standards and shall be certified by the manufacturer to ±1% of the tag value. Additionally, the manufacturer of the cylinder should provide a recommended shelf life for each calibration gas cylinder over which the concentration does not change more than ±2% from the certified value. For calibration gas values not generally available, alternative methods for preparing calibration gas mixtures, such as dilution systems, may be used with the approval of the Department.
2.1.9.1 Fuel. A 40% H2/60% He or 40% H2/60% N2 gas mixture is recommended to avoid an oxygen synergism effect that reportedly occurs when oxygen concentration varies significantly from a mean value.
2.1.9.2 Carrier Gas. High purity air with less than one ppm of organic material (as propane or carbon equivalent) or less than 0.1% of the span value, whichever is greater.
2.1.9.3 FIA Linearity Calibration Gases. Low-, mid-, and high-range gas mixture standards with nominal propane concentrations of 20-30, 45-55, and 70-80% of the span value in air, respectively. Other calibration values and other span values may be used if it can be shown to the Department's satisfaction that more accurate measurements would be achieved.
2.1.9.4 Dilution Check Gas. Gas mixture standard containing propane in air, approximately half the span value after dilution.
2.1.10 Particulate Filter. An in-stack or an out-of-stack glass fiber filter is recommended if exhaust gas particulate loading is significant. An out-of-stack filter must be heated to prevent any condensation unless it can be demonstrated that no condensation occurs.
2.2 Captured Emissions Volumetric Flow Rate.
2.2.1 Method 2 or 2A Apparatus. For determining volumetric flow rate.
2.2.2 Method 3 Apparatus and Reagents. For determining molecular weight of the gas stream. An estimate of the molecular weight of the gas stream may be used if approved by the Department.
2.2.3 Method 4 Apparatus and Reagents. For determining moisture content, if necessary.
3.0 Determination of Volumetric Flow Rate of Captured Emissions
3.1 Locate all points where emissions are captured from the affected facility. Using Method 1, determine the sampling points. Be sure to check each site for cyclonic or swirling flow.
3.2 Measure the velocity at each sampling site at least once every hour during each sampling run using Method 2 or 2A.
4.0 Determination of VOC Content of Captured Emissions
4.1 Analysis Duration. Measure the VOC responses at each captured emissions point during the entire test run or, if applicable, while the process is operating. If there are multiple captured emission locations, design a sampling system to allow a single FIA to be used to determine the VOC responses at all sampling locations.
4.2 Gas VOC Concentration.
4.2.1 Assemble the sample train. Calibrate the FIA according to the procedure in 5.1 of this method.
4.2.2 Conduct a system check according to the procedure in 5.3 of this method.
4.2.3 Install the sample probe so that the probe is centrally located in the stack, pipe, or duct, and is sealed tightly at the stack port connection.
4.2.4 Inject zero gas at the calibration valve assembly. Allow the measurement system response to reach zero. Measure the system response time as the time required for the system to reach the effluent concentration after the calibration valve has been returned to the effluent sampling position.
4.2.5 Conduct a system check before, and a system drift check after, each sampling run according to the procedures in 5.2 and 5.3 of this method. If the drift check following a run indicates unacceptable performance (see 5.3 of this method), the run is not valid. The tester may elect to perform system drift checks during the run not to exceed one drift check per hour.
4.2.6 Verify that the sample lines, filter, and pump temperatures are 120 ± 5°C.
4.2.7 Begin sampling at the start of the test period and continue to sample during the entire run. Record the starting and ending times and any required process information as appropriate. If multiple captured emission locations are sampled using a single FIA, sample at each location for the same amount of time (e.g., two minutes) and continue to switch from one location to another for the entire test run. Be sure that total sampling time at each location is the same at the end of the test run. Collect at least four separate measurements from each sample point during each hour of testing. Disregard the measurements at each sampling location until two times the response time of the measurement system has elapsed. Continue sampling for at least one minute and record the concentration measurements.
4.3 Background Concentration. NOTE: Not applicable when the building is used as the TTE.
4.3.1 Locate all NDO's of the TTE. A sampling point shall be at the center of each NDO, unless otherwise specified by the Department. If there are more than six NDO's, choose six sampling points evenly spaced among the NDO's.
4.3.2 Assemble the sample train. Calibrate the FIA and conduct a system check according to the procedures in 5.1 and 5.3 of this method. NOTE: This sample train shall be separate from the sample train used to measure the captured emissions.
4.3.3 Position the probe at the sampling location.
4.3.4 Determine the response time, conduct the system check, and sample according to the procedures described in 4.2.4 through 4.2.7 of this method.
4.4 Alternative Procedure. The direct interface sampling and analysis procedure described in Section 7.2 of Method 18 may be used to determine the gas VOC concentration (see 1.1 of Appendix E of this regulation). The system must be designed to collect and analyze at least one sample every 10 minutes.
5.0 Calibration and Quality Assurance
5.1 FIA Calibration and Linearity Check. Make necessary adjustments to the air and fuel supplies for the FIA and ignite the burner. Allow the FIA to warm up for the period recommended by the manufacturer. Inject a calibration gas into the measurement system and adjust the back-pressure regulator to the value required to achieve the flow rates specified by the manufacturer. Inject the zero- and the high-range calibration gases and adjust the analyzer calibration to provide the proper responses. Inject the low- and mid-range gases and record the responses of the measurement system. The calibration and linearity of the system are acceptable if the responses for all four gases are within 5% of the respective gas values. If the performance of the system is not acceptable, repair or adjust the system and repeat the linearity check. Conduct a calibration and linearity check after assembling the analysis system and after a major change is made to the system.
5.2 Systems Drift Checks. Select the calibration gas that most closely approximates the concentration of the captured emissions for conducting the drift checks. Introduce the zero and calibration gases at the calibration valve assembly and verify that the appropriate gas flow rate and pressure are present at the FIA. Record the measurement system responses to the zero and calibration gases. The performance of the system is acceptable if the difference between the drift check measurement and the value obtained in 5.1 of this method is less than 3% of the span value. Conduct the system drift checks at the end of each run.
5.3 System Check. Inject the high-range calibration gas at the inlet of the sampling probe and record the response. The performance of the system is acceptable if the measurement system response is within 5% of the value obtained in 5.1 of this method for the high-range calibration gas. Conduct a system check before and after each test run.
5.4 Analysis Audit Procedure. Immediately before each test, analyze an audit cylinder as described in 5.2 of this method. The analysis audit must agree with the audit cylinder concentration within 10%.
6.0 Nomenclature
CBi = corrected average VOC concentration of background emissions at point i, ppm propane.
CB = average background concentration, ppm propane.
CGj = corrected average VOC concentration of captured emissions at point j, ppm propane.
CDH = average measured concentration for the drift check calibration gas, ppm propane.
CD0 = average system drift check concentration for zero concentration gas, ppm propane.
CH = actual concentration of the drift check calibration gas, ppm propane.
Ci = uncorrected average background VOC concentration measured at point i, ppm propane.
Cj = uncorrected average VOC concentration measured at point j, ppm propane.
QGj = average effluent volumetric flow rate corrected to standard conditions at captured emissions point j, m3/min.
0C = total duration of captured emissions.
7.0 Calculations
7.1 Total VOC Captured Emissions.
7.2 VOC Concentration of the Captured Emissions at Point j.
7.3 Background VOC Concentration at Point i.
7.4 Average Background Concentration.
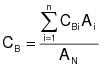 (D-30B-4)
(D-30B-4)NOTE: If the concentration at each point is within 20% of the average concentration of all points, then use the arithmetic average.
1.0 Introduction
1.1 Applicability. This procedure is applicable for determining the VOC content of captured gas streams. It is intended to be used in the development of a gas/gas protocol in which fugitive emissions are measured for determining VOC CE for surface coating and printing operations. A dilution system is used to reduce the VOC concentration of the captured emissions to about the same concentration as the fugitive emissions. The procedure may not be acceptable in certain site-specific situations [e.g., when: (1) direct-fired heaters or other circumstances affect the quantity of VOC at the control device inlet; and (2) particulate organic aerosols are formed in the process and are present in the captured emissions].
1.2 Principle. The amount of VOC captured (G) is calculated as the sum of the products of the VOC content (CGj), the flow rate (QGj), and the sampling time (0C) from each captured emissions point.
1.3 Estimated Measurement Uncertainty. The measurement uncertainties are estimated for each captured or fugitive emissions point as follows: QGj = ±5.5% and CGj = ±5%. Based on these numbers, the probable uncertainty for G is estimated at about ±7.4%.
1.4 Sampling Requirements. A CE test shall consist of at least three sampling runs. Each run shall cover at least one complete production cycle, but shall be at least three hours long. The sampling time for each run need not exceed eight hours, even if the production cycle has not been completed. Alternative sampling times may be used with the approval of the Department.
1.5 Notes. Because this procedure is often applied in highly explosive areas, caution and care should be exercised in choosing, installing, and using the appropriate equipment. Mention of trade names or company products does not constitute endorsement. All gas concentrations (percent, ppm) are by volume, unless otherwise noted.
2.0 Apparatus and Reagents
2.1 Gas VOC Concentration. The main components are as follows:
2.1.1 Dilution System. A Kipp in-stack dilution probe and controller or similar device may be used. The dilution rate may be changed by substituting different critical orifices or adjustments of the aspirator supply pressure. The dilution system shall be heated to prevent VOC condensation. Note: An out-of-stack dilution device may be used.
2.1.2 Calibration Valve Assembly. Three-way valve assembly at the outlet of the sample probe to direct the zero and calibration gases to the analyzer. Other methods, such as quick connect lines, to route calibration gases to the outlet of the sample probe are acceptable.
2.1.3 Sample Line. Stainless steel or Teflon tubing to transport the sample gas to the analyzer. The sample line must be heated to prevent condensation.
2.1.4 Sample Pump. A leak-free pump, to pull the sample gas through the system at a flow rate sufficient to minimize the response time of the measurement system. The components of the pump that contact the gas stream shall be constructed of stainless steel or Teflon. The sample pump must be heated to prevent condensation.
2.1.5 Sample Flow Rate Control. A sample flow rate control valve and rotameter, or equivalent, to maintain a constant sampling rate within 10%. The flow control valve and rotameter must be heated to prevent condensation. A control valve may also be located on the sample pump bypass loop to assist in controlling the sample pressure and flow rate.
2.1.6 Sample Gas Manifold. Capable of diverting a portion of the sample gas stream to the FIA, and the remainder to the bypass discharge vent. The manifold components shall be constructed of stainless steel or Teflon. If captured or fugitive emissions are to be measured at multiple locations, the measurement system shall be designed to use separate sampling probes, lines, and pumps for each measurement location and a common sample gas manifold and FIA. The sample gas manifold and connecting lines to the FIA must be heated to prevent condensation.
2.1.7 Organic Concentration Analyzer. An FIA with a span value of 1.5 times the expected concentration as propane; however, other span values may be used if it can be demonstrated to the Department's satisfaction that they would provide more accurate measurements. The system shall be capable of meeting or exceeding the following specifications:
2.1.7.1 Zero Drift. Less than ±3.0% of the span value.
2.1.7.2 Calibration Drift. Less than ±3.0% of the span value.
2.1.7.3 Calibration Error. Less than ±5.0% of the calibration gas value.
2.1.7.4 Response Time. Less than 30 seconds.
2.1.8 Integrator/Data Acquisition System. An analog or digital device or computerized data acquisition system used to integrate the FIA response or compute the average response and record measurement data. The minimum data sampling frequency for computing average or integrated values is one measurement value every five seconds. The device shall be capable of recording average values at least once per minute.
2.1.9 Calibration and Other Gases. Gases used for calibration, fuel, and combustion air (if required) are contained in compressed gas cylinders. All calibration gases shall be traceable to National Institute of Standards and Technology standards and shall be certified by the manufacturer to ±1% of the tag value. Additionally, the manufacturer of the cylinder should provide a recommended shelf life for each calibration gas cylinder over which the concentration does not change more than ±2% from the certified value. For calibration gas values not generally available, alternative methods for preparing calibration gas mixtures, such as dilution systems, may be used with the approval of the Department.
2.1.9.1 Fuel. A 40% H2/60% He or 40% H2/60% N2 gas mixture is recommended to avoid an oxygen synergism effect that reportedly occurs when oxygen concentration varies significantly from a mean value.
2.1.9.2 Carrier Gas and Dilution Air Supply. High purity air with less than one ppm of organic material (as propane or carbon equivalent), or less than 0.1% of the span value, whichever is greater.
2.1.9.3 FIA Linearity Calibration Gases. Low-, mid-, and high-range gas mixture standards with nominal propane concentrations of 20-30, 45-55, and 70-80% of the span value in air, respectively. Other calibration values and other span values may be used if it can be shown to the Department's satisfaction that more accurate measurements would be achieved.
2.1.9.4 Dilution Check Gas. Gas mixture standard containing propane in air, approximately half the span value after dilution.
2.1.10 Particulate Filter. An in-stack or an out-of-stack glass fiber filter is recommended if exhaust gas particulate loading is significant. An out-of-stack filter must be heated to prevent any condensation unless it can be demonstrated that no condensation occurs.
2.2 Captured Emissions Volumetric Flow Rate.
2.2.1 Method 2 or 2A Apparatus. For determining volumetric flow rate.
2.2.2 Method 3 Apparatus and Reagents. For determining molecular weight of the gas stream. An estimate of the molecular weight of the gas stream may be used if approved by the Department.
2.2.3 Method 4 Apparatus and Reagents. For determining moisture content, if necessary.
3.0 Determination of Volumetric Flow Rate of Captured Emissions
3.1 Locate all points where emissions are captured from the affected facility. Using Method 1, determine the sampling points. Be sure to check each site for cyclonic or swirling flow.
3.2 Measure the velocity at each sampling site at least once every hour during each sampling run using Method 2 or 2A.
4.0 Determination of VOC Content of Captured Emissions
4.1 Analysis Duration. Measure the VOC responses at each captured emissions point during the entire test run or, if applicable, while the process is operating. If there are multiple captured emissions locations, design a sampling system to allow a single FIA to be used to determine the VOC responses at all sampling locations.
4.2 Gas VOC Concentration.
4.2.1 Assemble the sample train. Calibrate the FIA according to the procedure in 5.1 of this method.
4.2.2 Set the dilution ratio and determine the dilution factor according to the procedure in 5.3 of this method.
4.2.3 Conduct a system check according to the procedure in 5.4 of this method.
4.2.4 Install the sample probe so that the probe is centrally located in the stack, pipe, or duct, and is sealed tightly at the stack port connection.
4.2.5 Inject zero gas at the calibration valve assembly. Measure the system response time as the time required for the system to reach the effluent concentration after the calibration valve has been returned to the effluent sampling position.
4.2.6 Conduct a system check before, and a system drift check after, each sampling run according to the procedures in 5.2 and 5.4 of this method. If the drift check following a run indicates unacceptable performance (see 5.4 of this method), the run is not valid. The tester may elect to perform system drift checks during the run not to exceed one drift check per hour.
4.2.7 Verify that the sample lines, filter, and pump temperatures are 120 ± 5°C.
4.2.8 Begin sampling at the start of the test period and continue to sample during the entire run. Record the starting and ending times and any required process information as appropriate. If multiple captured emission locations are sampled using a single FIA, sample at each location for the same amount of time (e.g., two min.) and continue to switch from one location to another for the entire test run. Be sure that total sampling time at each location is the same at the end of the test run. Collect at least four separate measurements from each sample point during each hour of testing. Disregard the measurements at each sampling location until two times the response time of the measurement system has elapsed. Continue sampling for at least one minute and record the concentration measurements.
4.3 Background Concentration. NOTE: Not applicable when the building is used as the TTE.
4.3.1 Locate all NDO's of the TTE. A sampling point shall be at the center of each NDO, unless otherwise approved by the Department. If there are more than six NDO's, choose six sampling points evenly spaced among the NDO's.
4.3.2 Assemble the sample train. Calibrate the FIA and conduct a system check according to the procedures in 5.1 and 5.4 of this method.
4.3.3 Position the probe at the sampling location.
4.3.4 Determine the response time, conduct the system check, and sample according to the procedures described in 4.2.4 through 4.2.8 of this method.
4.4 Alternative Procedure. The direct interface sampling and analysis procedure described in Section 7.2 of Method 18 may be used to determine the gas VOC concentration (see 1.1 of Appendix E of this regulation). The system must be designed to collect and analyze at least one sample every 10 minutes.
5.0 Calibration and Quality Assurance
5.1 FIA Calibration and Linearity Check. Make necessary adjustments to the air and fuel supplies for the FIA and ignite the burner. Allow the FIA to warm up for the period recommended by the manufacturer. Inject a calibration gas into the measurement system after the dilution system and adjust the back-pressure regulator to the value required to achieve the flow rates specified by the manufacturer. Inject the zero- and the high-range calibration gases and adjust the analyzer calibration to provide the proper responses. Inject the low- and mid-range gases and record the responses of the measurement system. The calibration and linearity of the system are acceptable if the responses for all four gases are within 5% of the respective gas values. If the performance of the system is not acceptable, repair or adjust the system and repeat the linearity check. Conduct a calibration and linearity check after assembling the analysis system and after a major change is made to the system.
5.2 Systems Drift Checks. Select the calibration gas that most closely approximates the concentration of the diluted captured emissions for conducting the drift checks. Introduce the zero and calibration gases at the calibration valve assembly, and verify that the appropriate gas flow rate and pressure are present at the FIA. Record the measurement system responses to the zero and calibration gases. The performance of the system is acceptable if the difference between the drift check measurement and the value obtained in 5.1 of this method is less than 3% of the span value. Conduct the system drift check at the end of each run.
5.3 Determination of Dilution Factor. Inject the dilution check gas into the measurement system before the dilution system and record the response. Calculate the dilution factor using Equation D-30C-3.
5.4 System Check. Inject the high-range calibration gas at the inlet to the sampling probe while the dilution air is turned off. Record the response. The performance of the system is acceptable if the measurement system response is within 5% of the value obtained in 5.1 of this method for the high-range calibration gas. Conduct a system check before and after each test run.
5.5 Analysis Audit Procedure. Immediately before each test, analyze an audit cylinder as described in 5.2 of this method. The analysis audit must agree with the audit cylinder concentration within 10%.
6.0 Nomenclature
CA = actual concentration of the dilution check gas, ppm propane.
CBi = corrected average VOC concentration of background emissions at point i, ppm propane.
CB = average background concentration, ppm propane.
CDH = average measured concentration for the drift check calibration gas, ppm propane.
CD0 = average system drift check concentration for zero concentration gas, ppm propane.
CH = actual concentration of the drift check calibration gas, ppm propane.
Cj = uncorrected average VOC concentration measured at point j, ppm propane.
CM = measured concentration of the dilution check gas, ppm propane.
DF = dilution factor.
QGj = average effluent volumetric flow rate corrected to standard conditions at captured emissions point j, m3/min.
0C = total duration of CE sampling run, min.
7.0 Calculations
7.1 Total VOC Captured Emissions.
7.2 VOC Concentration of the Captured Emissions at Point j.
7.3 Dilution Factor.
7.4 Background VOC Concentration at Point i.
7.5 Average Background Concentration.
 (D-30C-5)
(D-30C-5)1.0 Introduction
1.1 Applicability. This procedure is applicable for determining the fugitive VOC emissions from a TTE. It is intended to be used as a segment in the development of liquid/gas or gas/gas protocols for determining VOC CE for surface coating and printing operations.
1.2 Principle. The amount of fugitive VOC emissions (F) from the TTE is calculated as the sum of the products of the VOC content (CFj), the flow rate (QFj), and the sampling time (0F) from each fugitive emissions point.
1.3 Estimated Measurement Uncertainty. The measurement uncertainties are estimated for each fugitive emission point as follows:
QFj = ±5.5% and CFj = ±5.0%. Based on these numbers, the probable uncertainty for F is estimated at about ±7.4%.
1.4 Sampling Requirements. A CE test shall consist of at least three sampling runs. Each run shall cover at least one complete production cycle, but shall be at least three hours long. The sampling time for each run need not exceed eight hours, even if the production cycle has not been completed. Alternative sampling times may be used with the approval of the Department.
1.5 Notes. Because this procedure is often applied in highly explosive areas, caution and care should be exercised in choosing, installing, and using the appropriate equipment. Mention of trade names or company products does not constitute endorsement. All gas concentrations (percent, ppm) are by volume, unless otherwise noted.
2.0 Apparatus and Reagents
2.1 Gas VOC Concentration. The main components are as follows:
2.1.1 Sample Probe. Stainless steel or equivalent. The probe shall be heated to prevent VOC condensation.
2.1.2 Calibration Valve Assembly. Three-way valve assembly at the outlet of the sample probe to direct the zero and calibration gases to the analyzer. Other methods, such as quick-connect lines, to route calibration gases to the outlet of the sample probe are acceptable.
2.1.3 Sample Line. Stainless steel or Teflon tubing to transport the sample gas to the analyzer. The sample line must be heated to prevent condensation.
2.1.4 Sample Pump. A leak-free pump, to pull the sample gas through the system at a flow rate sufficient to minimize the response time of the measurement system. The components of the pump that contact the gas stream shall be constructed of stainless steel or Teflon. The sample pump must be heated to prevent condensation.
2.1.5 Sample Flow Rate Control. A sample flow rate control valve and rotameter, or equivalent, to maintain a constant sampling rate within 10%. The flow control valve and rotameter must be heated to prevent condensation. A control valve may also be located on the sample pump bypass loop to assist in controlling the sample pressure and flow rate.
2.1.6 Sample Gas Manifold. Capable of diverting a portion of the sample gas stream to the FIA, and the remainder to the bypass discharge vent. The manifold components shall be constructed of stainless steel or Teflon. If emissions are to be measured at multiple locations, the measurement system shall be designed to use separate sampling probes, lines, and pumps for each measurement location and a common sample gas manifold and FIA. The sample gas manifold and connecting lines to the FIA must be heated to prevent condensation.
2.1.7 Organic Concentration Analyzer. An FIA with a span value of 1.5 times the expected concentration as propane; however, other span values may be used if it can be demonstrated to the Department's satisfaction that they would provide more accurate measurements. The system shall be capable of meeting or exceeding the following specifications:
2.1.7.1 Zero Drift. Less than ±3.0% of the span value.
2.1.7.2 Calibration Drift. Less than ±3.0% of the span value.
2.1.7.3 Calibration Error. Less than ±5.0% of the calibration gas value.
2.1.7.4 Response Time. Less than 30 seconds.
2.1.8 Integrator/Data Acquisition System. An analog or digital device or computerized data acquisition system used to integrate the FIA response or compute the average response and record measurement data. The minimum data sampling frequency for computing average or integrated values is one measurement value every five seconds. The device shall be capable of recording average values at least once per minute.
2.1.9 Calibration and Other Gases. Gases used for calibration, fuel, and combustion air (if required) are contained in compressed gas cylinders. All calibration gases shall be traceable to National Institute of Standards and Technology standards and shall be certified by the manufacturer to ±1% of the tag value. Additionally, the manufacturer of the cylinder should provide a recommended shelf life for each calibration gas cylinder over which the concentration does not change more than ±2% from the certified value. For calibration gas values not generally available, alternative methods for preparing calibration gas mixtures, such as dilution systems, may be used with the approval of the Department.
2.1.9.1 Fuel. A 40% H2/60% He or 40% H2/60% N2 gas mixture is recommended to avoid an oxygen synergism effect that reportedly occurs when oxygen concentration varies significantly from a mean value.
2.1.9.2 Carrier Gas. High purity air with less than one ppm of organic material (as propane or carbon equivalent) or less than 0.1% of the span value, whichever is greater.
2.1.9.3 FIA Linearity Calibration Gases. Low-, mid-, and high-range gas, mixture standards with nominal propane concentrations of 20-30, 45-55, and 70-80% of the span value in air, respectively. Other calibration values and other span values may be used if it can be shown to the Department's satisfaction that more accurate measurements would be achieved.
2.1.10 Particulate Filter. An in-stack or an out-of-stack glass fiber filter is recommended if exhaust gas particulate loading is significant. An out-of-stack filter must be heated to prevent any condensation unless it can be demonstrated that no condensation occurs.
2.2 Fugitive Emissions Volumetric Flow Rate.
2.2.1 Method 2 or 2A Apparatus. For determining volumetric flow rate.
2.2.2 Method 3 Apparatus and Reagents. For determining molecular weight of the gas stream. An estimate of the molecular weight of the gas stream may be used if approved by the Department.
2.2.3 Method 4 Apparatus and Reagents. For determining moisture content, if necessary.
2.3 Temporary Total Enclosure. The criteria for designing an acceptable TTE are specified in Method 30.
3.0 Determination of Volumetric Flow Rate of Fugitive Emissions
3.1 Locate all points where emissions are exhausted from the TTE. Using Method 1, determine the sampling points. Be sure to check each site for cyclonic or swirling flow.
3.2 Measure the velocity at each sampling site at least once every hour during each sampling run using Method 2 or 2A.
4.0 Determination of VOC Content of Fugitive Emissions
4.1 Analysis Duration. Measure the VOC responses at each fugitive emission point during the entire test run or, if applicable, while the process is operating. If there are multiple emission locations, design a sampling system to allow a single FIA to be used to determine the VOC responses at all sampling locations.
4.2 Gas VOC Concentration.
4.2.1 Assemble the sample train. Calibrate the FIA and conduct a system check according to the procedures in 5.1 and 5.3 of this method, respectively.
4.2.2 Install the sample probe so that the probe is centrally located in the stack, pipe, or duct, and is sealed tightly at the stack port connection.
4.2.3 Inject zero gas at the calibration valve assembly. Allow the measurement system response to reach zero. Measure the system response time as the time required for the system to reach the effluent concentration after the calibration valve has been returned to the effluent sampling position.
4.2.4 Conduct a system check before, and a system drift check after, each sampling run according to the procedures in 5.2 and 5.3 of this method. If the drift check following a run indicates unacceptable performance (see 5.3 of this method), the run is not valid. The tester may elect to perform system drift checks during the run not to exceed one drift check per hour.
4.2.5 Verify that the sample lines, filter, and pump temperatures are 120 ± 5°C.
4.2.6 Begin sampling at the start of the test period and continue to sample during the entire run. Record the starting and ending times and any required process information, as appropriate. If multiple emission locations are sampled using a single FIA, sample at each location for the same amount of time (e.g., two min.) and continue to switch from one location to another for the entire test run. Be sure that total sampling time at each location is the same at the end of the test run. Collect at least four separate measurements from each sample point during each hour of testing. Disregard the response measurements at each sampling location until two times the response time of the measurement system has elapsed. Continue sampling for at least one minute and record the concentration measurements.
4.3 Background Concentration.
4.3.1 Locate all NDO's of the TTE. A sampling point shall be at the center of each NDO, unless otherwise approved by the Department. If there are more than six NDO's, choose six sampling points evenly spaced among the NDO's.
4.3.2 Assemble the sample train. Calibrate the FIA and conduct a system check according to the procedures in 5.1 and 5.3 of this method.
4.3.3 Position the probe at the sampling location.
4.3.4 Determine the response time, conduct the system check, and sample according to the procedures described in 4.2.3 through 4.2.6 of this method.
4.4 Alternative Procedure. The direct interface sampling and analysis procedure described in Section 7.2 of Method 18 may be used to determine the gas VOC concentration (see 1.1 of Appendix E of this regulation). The system must be designed to collect and analyze at least one sample every 10 minutes.
5.0 Calibration and Quality Assurance
5.1 FIA Calibration and Linearity Check. Make necessary adjustments to the air and fuel supplies for the FIA and ignite the burner. Allow the FIA to warm up for the period recommended by the manufacturer. Inject a calibration gas into the measurement system and adjust the back-pressure regulator to the value required to achieve the flow rates specified by the manufacturer. Inject the zero- and the high-range calibration gases and adjust the analyzer calibration to provide the proper responses. Inject the low- and mid-range gases and record the responses of the measurement system. The calibration and linearity of the system are acceptable if the responses for all four gases are within 5% of the respective gas values. If the performance of the system is not acceptable, repair or adjust the system and repeat the linearity check. Conduct a calibration and linearity check after assembling the analysis system and after a major change is made to the system.
5.2 Systems Drift Checks. Select the calibration gas concentration that most closely approximates that of the fugitive gas emissions to conduct the drift checks. Introduce the zero and calibration gases at the calibration valve assembly and verify that the appropriate gas flow rate and pressure are present at the FIA. Record the measurement system responses to the zero and calibration gases. The performance of the system is acceptable if the difference between the drift check measurement and the value obtained in 5.1 of this method is less than 3% of the span value. Conduct a system drift check at the end of each run.
5.3 System Check. Inject the high-range calibration gas at the inlet of the sampling probe and record the response. The performance of the system is acceptable if the measurement system response is within 5% of the value obtained in 5.1 of this method for the high-range calibration gas. Conduct a system check before each test run.
5.4 Analysis Audit. Immediately before each test, analyze an audit cylinder as described in 5.2 of this method. The analysis audit must agree with the audit cylinder concentration within 10%.
6.0 Nomenclature
CBi = corrected average VOC concentration of background emissions at point i, ppm propane.
CB = average background concentration, ppm propane.
CDH = average measured concentration for the drift check calibration gas, ppm propane.
CD0 = average system drift check concentration for zero concentration gas, ppm propane.
CFj = corrected average VOC concentration of fugitive emissions at point j, ppm propane.
CH = actual concentration of the drift check calibration gas, ppm propane.
Ci = uncorrected average background VOC concentration at point i, ppm propane.
Cj = uncorrected average VOC concentration measured at point j, ppm propane.
QFj = average effluent volumetric flow rate corrected to standard conditions at fugitive emissions point j, m3/min.
0F = total duration of fugitive emissions sampling run, min.
7.0 Calculations
7.1 Total VOC Fugitive Emissions.
7.2 VOC Concentration of the Fugitive Emissions at Point j.
7.3 Background VOC Concentration at Point i.
7.4 Average Background Concentration.
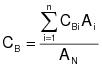 (D-30D-4)
(D-30D-4)NOTE: If the concentration at each point is within 20% of the average concentration of all points, use the arithmetic average.
1.0 Introduction
1.1 Applicability. This procedure is applicable for determining the fugitive VOC emissions from a BE. It is intended to be used in the development of liquid/gas or gas/gas protocols for determining VOC CE for surface coating and printing operations.
1.2 Principle. The total amount of fugitive VOC emissions (FB) from the BE is calculated as the sum of the products of the VOC content (CFj) of each fugitive emissions point, the flow rate (QFj) at each fugitive emissions point, and time (0F).
1.3 Measurement Uncertainty. The measurement uncertainties are estimated for each fugitive emissions point as follows: QFj = ±10.0% and CFj = ±5.0%. Based on these numbers, the probable uncertainty for FB is estimated at about ±11.2%.
1.4 Sampling Requirements. A CE test shall consist of at least three sampling runs. Each run shall cover at least one complete production cycle, but shall be at least three hours long. The sampling time for each run need not exceed eight hours, even if the production cycle has not been completed. Alternative sampling times may be used with the approval of the Department.
1.5 Notes. Because this procedure is often applied in highly explosive areas, caution and care should be exercised in choosing, installing, and using the appropriate equipment. Mention of trade names or company products does not constitute endorsement. All gas concentrations (percent, ppm) are by volume, unless otherwise noted.
2.0 Apparatus and Reagents
2.1 Gas VOC Concentration. The main components are as follows:
2.1.1 Sample Probe. Stainless steel or equivalent. The probe shall be heated to prevent VOC condensation.
2.1.2 Calibration Valve Assembly. Three-way valve assembly at the outlet of the sample probe to direct the zero and calibration gases to the analyzer. Other methods, such as quick-connect lines, to route calibration gases to the outlet of the sample probe are acceptable.
2.1.3 Sample Line. Stainless steel or Teflon tubing to transport the sample gas to the analyzer. The sample line must be heated to prevent condensation.
2.1.4 Sample Pump. A leak-free pump, to pull the sample gas through the system at a flow rate sufficient to minimize the response time of the measurement system. The components of the pump that contact the gas stream shall be constructed of stainless steel or Teflon. The sample pump must be heated to prevent condensation.
2.1.5 Sample Flow Rate Control. A sample flow rate control valve and rotameter, or equivalent, to maintain a constant sampling rate within 10%. The flow rate control valve and rotameter must be heated to prevent condensation. A control valve may also be located on the sample pump bypass loop to assist in controlling the sample pressure and flow rate.
2.1.6 Sample Gas Manifold. Capable of diverting a portion of the sample gas stream to the FIA, and the remainder to the bypass discharge vent. The manifold components shall be constructed of stainless steel or Teflon. If emissions are to be measured at multiple locations, the measurement system shall be designed to use separate sampling probes, lines, and pumps for each measurement location, and a common sample gas manifold and FIA. The sample gas manifold must be heated to prevent condensation.
2.1.7 Organic Concentration Analyzer. An FIA with a span value of 1.5 times the expected concentration as propane; however, other span values may be used if it can be demonstrated to the Department's satisfaction that they would provide more accurate measurements. The system shall be capable of meeting or exceeding the following specifications:
2.1.7.1 Zero Drift. Less than ±3.0% of the span value.
2.1.7.2 Calibration Drift. Less than ±3.0% of the span value.
2.1.7.3 Calibration Error. Less than ±5.0% of the calibration gas value.
2.1.7.4 Response Time. Less than 30 seconds.
2.1.8 Integrator/Data Acquisition System. An analog or digital device or computerized data acquisition system used to integrate the FIA response or compute the average response and record measurement data. The minimum data sampling frequency for computing average or integrated values is one measurement value every 5 seconds. The device shall be capable of recording average values at least once per minute.
2.1.9 Calibration and Other Gases. Gases used for calibration, fuel, and combustion air (if required) are contained in compressed gas cylinders. All calibration gases shall be traceable to National Institute of Standards and Technology standards and shall be certified by the manufacturer to ±1% of the tag value. Additionally, the manufacturer of the cylinder should provide a recommended shelf life for each calibration gas cylinder over which the concentration does not change more than ±2% from the certified value. For calibration gas values not generally available, alternative methods for preparing calibration gas mixtures, such as dilution systems, may be used with the approval of the Department.
2.1.9.1 Fuel. A 40% H2/60% He or 40% H2/60% N2 gas mixture is recommended to avoid an oxygen synergism effect that reportedly occurs when oxygen concentration varies significantly from a mean value.
2.1.9.2 Carrier Gas. High purity air with less than one ppm of organic material (propane or carbon equivalent) or less than 0.1% of the span value, whichever is greater.
2.1.9.3 FIA Linearity Calibration Gases. Low-, mid-, and high-range gas mixture standards with nominal propane concentrations of 20-30, 45-55, and 70-80% of the span value in air, respectively. Other calibration values and other span values may be used if it can be shown to the Department's satisfaction that more accurate measurements would be achieved.
2.1.10 Particulate Filter. An in-stack or an out-of-stack glass fiber filter is recommended if exhaust gas particulate loading is significant. An out-of-stack filter must be heated to prevent any condensation unless it can be demonstrated that no condensation occurs.
2.2 Fugitive Emissions Volumetric Flow Rate.
2.2.1 Flow Direction Indicators. Any means of indicating inward or outward flow, such as light plastic film or paper streamers, smoke tubes, filaments, and sensory perception.
2.2.2 Method 2 or 2A Apparatus. For determining volumetric flow rate. Anemometers or similar devices calibrated according to the manufacturer's instructions may be used when low velocities are present. Vane anemometers (Young-maximum response propeller), specialized pitots with electronic manometers (e.g., Shortridge Instruments Inc., Airdata Multimeter 860) are commercially available with measurement thresholds of 15 and eight mpm (50 and 25 fpm), respectively.
2.2.3 Method 3 Apparatus and Reagents. For determining molecular weight of the gas stream. An estimate of the molecular weight of the gas stream may be used if approved by the Department.
2.2.4 Method 4 Apparatus and Reagents. For determining moisture content, if necessary.
2.3 Total Temporary Enclosure. The criteria for an acceptable TTE are specified in Method 30.
3.0 Determination of Volumetric Flow Rate of Fugitive Emissions
3.1 Preliminary Determinations. To determine which exhaust points should be measured for volumetric flow rates and VOC concentrations, the following procedure shall be used:
3.1.1 Forced Draft Openings. Identify all forced draft openings. Determine the volumetric flow rate according to Method 2.
3.1.2 The NDO's Exhaust Points. The NDO's in the roof of a facility are considered to be exhaust points. Determine volumetric flow rate from these NDO's. Divide the cross-sectional area according to Method 1 using 12 equal areas. Use the appropriate velocity measurement devices (e.g., propeller anemometers).
3.1.3 Other NDO's.
3.1.3.1 This step is optional. Determine the exhaust flow rate, including that of the control device, from the enclosure and the intake air flow rate. If the exhaust flow rate divided by the intake air flow rate is greater than 1.1, then all other NDO's are not considered to be significant exhaust points. It is not necessary to measure the volumetric flow rate and VOC concentration from these insignificant exhaust points during the CE test.
3.1.3.2 If the option above is not taken, identify all other NDO's and other potential points through which fugitive emissions may escape the enclosure; then use the following criteria to determine whether flow rates and VOC concentrations need to be measured.
3.1.3.2.1 Using the appropriate flow direction indicator, determine the flow direction. An NDO with zero or inward flow is not an exhaust point.
3.1.3.2.2 Measure the outward volumetric flow rate from the remainder of the NDO's. If the collective flow rate is 2% or less of the flow rate from 3.1.1 and 3.1.2 of this method, then these NDO's, except those within two equivalent diameters (based on NDO opening) from VOC sources, may be considered to be non-exhaust points.
3.1.3.2.3 If the percentage calculated in 3.1.3.2.2 of this method is greater than 2%, those NDO's (except those within two equivalent diameters from VOC sources) whose volumetric flow rates total 2% of the flow rate from 3.1.1 and 3.1.2 of this method may be considered as non-exhaust points. All remaining NDO's shall be measured for volumetric flow rate and VOC concentrations during the CE test.
3.1.3.2.4 The tester may choose to measure VOC concentrations at the forced exhaust points and the NDO's. If the total VOC emissions from the non-roof NDO's are less than 2% of the emissions from the forced draft and roof NDO's, then it is not necessary to measure the VOC concentration at the non-roof NDO's during the CE test.
3.2 Determination of Flow Rates.
3.2.1 Measure the volumetric flow rate at all locations identified as exhaust points in 3.1 in this method. Divide each exhaust opening into nine equal areas for rectangular openings and into eight equal areas for circular openings.
3.2.2 Measure the velocity at each site at least once every hour during each sampling run using Method 2 or 2A, if applicable, or using the low velocity instruments in 2.2.2 in this method.
4.0 Determination of VOC Content of Fugitive Emissions
4.1 Analysis Duration. Measure the VOC responses at each fugitive emissions point during the entire test run or, if applicable, while the process is operating. If there are multiple emissions locations, design a sampling system to allow a single FIA to be used to determine the VOC responses at all sampling locations.
4.2 Gas VOC Concentration.
4.2.1 Assemble the sample train. Calibrate the FIA and conduct a system check according to the procedures in 5.1 and 5.3 in this method, respectively.
4.2.2 Install the sample probe so that the probe is centrally located in the stack, pipe, or duct, and is sealed tightly at the stack port connection.
4.2.3 Inject zero gas at the calibration valve assembly. Allow the measurement system response to reach zero. Measure the system response time as the time required for the system to reach the effluent concentration after the calibration valve has been returned to the effluent sampling position.
4.2.4 Conduct a system check before, and a system drift check after, each sampling run according to the procedures in 5.2 and 5.3 of this method. If the drift check following a run indicates unacceptable performance (see 5.3 of this method), the run is not valid. The tester may elect to perform drift checks during the run, not to exceed one drift check per hour.
4.2.5 Verify that the sample lines, filter, and pump temperatures are 120 ± 5°C.
4.2.6 Begin sampling at the start of the test period and continue to sample during the entire run. Record the starting and ending times, and any required process information, as appropriate. If multiple emission locations are sampled using a single FIA, sample at each location for the same amount of time (e.g., two minutes) and continue to switch from one location to another for the entire test run. Be sure that total sampling time at each location is the same at the end of the test run. Collect at least four separate measurements from each sample point during each hour of testing. Disregard the response measurements at each sampling location until two times the response time of the measurement system has elapsed. Continue sampling for at least one minute, and record the concentration measurements.
4.3 Alternative Procedure. The direct interface sampling and analysis procedure described in Section 7.2 of Method 18 may be used to determine the gas VOC concentration (see 1.1 of Appendix E of this regulation). The system must be designed to collect and analyze at least one sample every 10 minutes.
5.0 Calibration and Quality Assurance
5.1 FIA Calibration and Linearity Check. Make necessary adjustments to the air and fuel supplies for the FIA and ignite the burner. Allow the FIA to warm up for the period recommended by the manufacturer. Inject a calibration gas into the measurement system and adjust the back-pressure regulator to the value required to achieve the flow rates specified by the manufacturer. Inject the zero- and the high-range calibration gases, and adjust the analyzer calibration to provide the proper responses. Inject the low- and mid-range gases and record the responses of the measurement system. The calibration and linearity of the system are acceptable if the responses for all four gases are within 5% of the respective gas values. If the performance of the system is not acceptable, repair or adjust the system and repeat the linearity check. Conduct a calibration and linearity check after assembling the analysis system and after a major change is made to the system.
5.2 Systems Drift Checks. Select the calibration gas that most closely approximates the concentration of the captured emissions for conducting the drift checks. Introduce the zero and calibration gases at the calibration valve assembly and verify that the appropriate gas flow rate and pressure are present at the FIA. Record the measurement system responses to the zero and calibration gases. The performance of the system is acceptable if the difference between the drift check measurement and the value obtained in 5.1 of this method is less than 3% of the span value. Conduct a system drift check at the end of each run.
5.3 System Check. Inject the high-range calibration gas at the inlet of the sampling probe and record the response. The performance of the system is acceptable if the measurement system response is within 5% of the value obtained in 5.1 of this method for the high-range calibration gas. Conduct a system check before each test run.
5.4 Analysis Audit. Immediately before each test, analyze an audit cylinder as described in 5.2 of this method. The analysis audit must agree with the audit cylinder concentration within 10%.
6.0 Nomenclature
CDH = average measured concentration for the drift check calibration gas, ppm propane.
CD0 = average system drift check concentration for zero concentration gas, ppm propane.
CFj = corrected average VOC concentration of fugitive emissions at point j, ppm propane.
CH = actual concentration of the drift check calibration gas, ppm propane.
Cj = uncorrected average VOC concentration measured at point j, ppm propane.
FB = total VOC content of fugitive emissions from the building, kg.
QFj = average effluent volumetric flow rate corrected to standard conditions at fugitive emissions point j, m3/min.
0F = total duration of CE sampling run, min.
7.0 Calculations
7.1 Total VOC Fugitive Emissions from the Building.
7.2 VOC Concentration of the Fugitive Emissions at Point j.
1.0 Depending upon the conditions at a test site, one of the following test methods from 40 CFR Part 60, Appendix A (July 1, 1992), must be used to determine volatile organic compound (VOC) concentrations of a gas stream at the inlet and outlet of a control device:
1.1 Method 18.
1.2 Method 25.
1.3 Method 25A.
2.0 The method selected shall be based on consideration of the diversity of organic species present and their total concentration and on consideration of the potential presence of interfering gases. Because of the different response factors for the many organic compounds formed during the combustion process, only Method 25, which measures VOC as carbon, shall be used for determining destruction efficiency of incinerators or catalytic incinerators, except in cases where the allowable outlet VOC concentration of the control device is less than 50 ppm as carbon, or the product of CO2 in percent and H2O in percent exceeds 100, in which case Method 25A shall be used.
3.0 Except as indicated in 3.1 and 3.2 of this appendix, a test shall consist of three separate runs, each lasting a minimum of 60 minutes (min), unless the Department determines that process variables dictate shorter sampling times.
3.1 When the method is to be used to determine the efficiency of a fixed-bed carbon adsorption system with a common exhaust stack for all of the individual adsorber vessels, the test shall consist of three separate runs, each coinciding with one or more complete sequences through the adsorption cycles of all the individual adsorber vessels.
3.2 When the method is to be used to determine the efficiency of a fixed-bed carbon adsorption system with individual exhaust stacks for each adsorber vessel, each adsorber vessel shall be tested individually. The test for each adsorber vessel shall consist of three separate runs. Each run shall coincide with one or more complete adsorption cycles.
4.0 Method 1 or 1A of 40 CFR Part 60, Appendix A (July 1, 1992), shall be used for velocity traverses.
5.0 Method 2, 2A, 2C, or 2D of 40 CFR Part 60, Appendix A (July 1, 1992), shall be used for velocity and volumetric flow rates.
6.0 Method 3 or 3A of 40 CFR Part 60, Appendix A (July 1, 1992), shall be used for O2 and CO2 analysis.
7.0 Method 4 of 40 CFR Part 60, Appendix A (July 1, 1992), shall be used for stack gas moisture.
8.0 Methods 2, 2A, 2C, 2D, 3, 3A and 4 of 40 CFR Part 60, Appendix A (July 1, 1992), shall be performed, as applicable, at least twice during each test run.
9.0 Use of adaptations to test methods. Use of an adaptation to any of the analytical methods specified in 1.0 and 4.0 through 8.0 of this appendix shall be approved by the Department on a case-by-case basis. An owner or operator shall submit sufficient documentation for the Department to find that the analytical methods specified in 1.0 and 4.0 through 8.0 of this appendix will yield inaccurate results and that the proposed adaptation is appropriate.
1.0 Owners or operators required to carry out a leak detection monitoring program shall comply with the following requirements:
1.1 Monitoring shall be performed in accordance with Method 21 of 40 CFR, Part 60, Appendix A (July 1, 1992).
1.2 The detection instrument shall meet the performance criteria of Method 21.
1.3 The detection instrument shall be calibrated before and after use on each day of its use by the methods specified in Method 21. Failure to achieve a post-use calibration precision of less than 10% shall constitute grounds for rejecting all tests performed since the last pre-use calibration. In such cases, required leak tests must be reperformed.
1.4 Calibration gases shall be:
1.4.1 Zero air (less than 10 parts per million [ppm] of hydrocarbon in air).
1.4.2 A mixture of methane or n-hexane and air at a concentration of approximately, but less than, 10,000 ppm methane or n-hexane.
1.5 The detection instrument probe shall be traversed around all potential leak interfaces as close to the interface as possible as described in Method 21.
2.0 When equipment is tested for compliance with the requirement that there be no detectable emissions, the test shall comply with the following:
2.1 The requirements of 1.1 through 1.5 of this appendix shall apply and shall be met, and
2.2 The background level shall be determined as set forth in Method 21.
3.0 Leak detection tests shall be performed consistent with:
3.1 "APTI Course SI 417-Controlling Volatile Organic Compound Emissions from Leaking Process Equipment", EPA-450/2-82-015.
3.2 "Portable Instrument User's Manual for Monitoring VOC Sources," EPA-340/1-86-015.
3.3 "Protocols for Generating Unit--Specific Emission Estimates for Equipment Leaks of VOC and VHAP," EPA-450/3-88-010.
3.4 "Petroleum Refinery Enforcement Manual", EPA-340/1-80-008.
4.0 Use of adaptations to test methods. Use of an adaptation to any of the analytical methods specified in 1.0, 2.0, and 3.0 of this appendix shall be approved by the Department on a case-by-case basis. An owner or operator shall submit sufficient documentation for the Department to find that the analytical methods specified in 1.0, 2.0, and 3.0 of this appendix will yield inaccurate results and that the proposed adaptation is appropriate.
1.0 Applicability.
1.1 This method applies to the measurement of total hydrocarbons as a surrogate measure for the total gaseous organic concentration of the VOC control device outlet. The concentration is expressed in terms of propane.
1.2 The Department may approve the use of gas conditioning, including cooling to between 4.4 and 18°C (40° and 64°F), and condensate traps to reduce the moisture content of the sample gas if the owner/operator:
1.2.1 Successfully demonstrates to the Department that the use of such system is necessary for the specific application.
1.2.2 Includes in the demonstration a quantification of the total hydrocarbon concentration (THC) lost to the gas conditioning system.
2.0 Principal.
A gas sample is extracted from the source through a heated sample line and heated glass fiber filter to a flame ionization detector (FID), or other detector as approved by the Department. Results are reported as volume concentration equivalents of the propane.
3.0 Definitions.
As used in this appendix, all terms not defined herein shall have the meaning given them in the November 15, 1990 Clean Air Act Amendments (CAAA), or in 2.0 of this regulation.
“Calibration drift” means the difference in the measurement system response to a mid-level calibration gas before and after a stated period of operation during which no unscheduled maintenance, repair, or adjustment took place.
“Calibration error” means the difference between the gas concentration indicated by the measurement system and the known concentration of the calibration system.
“Measurement system” means the total equipment required for the determination of the inlet and outlet gas concentrations, percent capture efficiency, and gas outlet emission rate. The system consists of the following major subsystems:
1. Sample interface--the portion of the system that is used for one or more of the following:
i. Sample acquisition.
ii. Sample transportation.
iii. Sample conditioning.
iv. Protection of the analyzer from the effects of the stack effluent.
2. Organic analyzer--the portion of the system that senses organic concentration and generates an output proportional to the gas concentration.
3. Data recorder--the portion of the system that records a permanent record of the measurement values.
4. Flow rate system
i. A gas volume meter meeting the requirements of Method 2A, Section 2.1 (40 CFR Part 60, Appendix A, July 1, 1992), or,
ii. A system approved by the Department.
“Response time” means the time interval from a step change in pollutant concentration at the inlet to the emission measurement system to the time at which 95% of the corresponding final value is reached as displayed on the recorder.
“Span value” means for most incinerators, a 50 parts per million (ppm) propane span. Different span values may be allowed with prior Department approval.
“Zero drift” means the difference in the measurement system response to a zero level calibration gas before and after a stated period of operation during which no unscheduled maintenance, repair, or adjustment took place.
4.0 Apparatus.
[Note: this method is often applied in highly explosive areas. Caution should be exercised in choice of equipment and installation.] An acceptable measurement system includes a sample interface system, a calibration valve, gas filter and a pump preceding the analyzer. THC measurement systems are designated HOT or COLD systems based on the operating temperatures of the system. In HOT systems, all components in contact with the sample gas (probe, calibration valve, filter, and sample lines) as well as all parts of the flame ionization analyzer between the sample inlet and the detector must be maintained between 150° to 175°C. This includes the sample pump if it is located on the inlet side of the FID. A condensate trap may be installed, if necessary, to prevent any condensate entering the detector. The essential components of the measurement system are as follows:
4.1 Organic concentration analyzer. A Department-approved detector capable of meeting or exceeding the specifications in this method.
4.2 Sample probe. Use either 4.2.1 or 4.2.2 of this appendix.
4.2.1 Stainless steel, or equivalent, three-hole rake type. Sample holes shall be four millimeters (mm) (0.2 inches [in.] in diameter or smaller and located at 16.7, 50, and 83.3% of the equivalent stack diameter.
4.2.2 A single opening probe so that a gas sample is collected from the centrally located 10% area of the stack cross-section, and is representative of the emission.
4.3 Sample line. Stainless steel or Teflon1 tubing to transport the sample gas to the analyzer. The sample line from the heated probe shall be heated to between 150 and 175°C (302 and 347°F).
4.4 Calibration valve assembly. Use either 4.4.1 or 4.4.2 of this appendix.
4.4.1 A heated three-way valve assembly to direct the zero and calibration gases to the analyzers.
4.4.2 Other methods, such as quick-connect lines, to route calibration gas to the analyzers.
4.5 Particulate filter. An in-stack or an out-of-stack glass fiber filter if exhaust gas particulate loading is significant. An out-of-stack filter must be heated.
4.6 Recorder. A strip-chart recorder, analog computer, or digital recorder for recording measurement data. The minimum data recording shall be one measurement value per minute. Full scale of the data recorder shall be selected so that the emission limit is between 30% and 50%.
5.0 Calibration gases and other gases.
5.1 Gases used for calibration, fuel, and combustion air shall be contained in compressed gas cylinders.
5.2 Preparation of calibration gases shall be done according to the procedure in Protocol No. 1, listed in the reference in 12.2 of this appendix.
5.3 The recommended shelf life for each calibration gas cylinder over which the concentration does not change more than ±2% from the certified value shall be obtained from the cylinder manufacturer.
5.4 The following calibration and other gases shall be used:
5.4.1 Fuel. A 40% hydrogen and 60% helium or 40% hydrogen and 60% nitrogen gas mixture to avoid an oxygen synergism effect that reportedly occurs when oxygen concentration varies significantly from a mean value.
5.4.2 Zero gas. High purity air with less than 0.1 parts per million by volume (ppmv) of organic material methane or carbon equivalent or less than 0.1% of the span value, whichever is greater.
5.4.3 Low-level calibration gas. Propane calibration gas (in air or nitrogen) with a concentration equivalent to 20 to 30% of the applicable span value.
5.4.4 Mid-level calibration gas. Propane calibration gas with a concentration equivalent to 45 to 55% of the applicable span value.
5.4.5 High-level calibration gas. Propane calibration gas with a concentration equivalent to 80 to 90% of the applicable span value.
6.0 Measurement system performance specifications.
6.1 Zero drift shall be less than ±3% of the span value.
6.2 Calibration drift shall be less than ±3% of the span value.
6.3 Calibration error shall be less than ±5% of the calibration gas value.
7.0 Pretest preparations.
7.1 Selection of sampling site.
7.1.1 The location of the sampling site shall be determined from the applicable provisions of this regulation or purpose of the test (i.e., exhaust stack, inlet line, etc).
7.1.2 The sample port shall be located at least 1.5 meters (4.9 feet) or two equivalent diameters upstream of the gas discharge to the atmosphere.
7.2 Location of sample probe. The sample probe must be installed so that the probe is centrally located in the stack, pipe or duct and is sealed tightly at the stack port connection.
7.3 Measurement systems preparation. Prior to the emission test, the measurement system must be assembled following the manufacturer's written instructions in preparing the sample interface and the organic analyzer. The system must be operable.
7.4 Calibration error test.
7.4.1 Immediately prior to the test series (within two hours of the start of the test), zero gas and high-level calibration gas shall be introduced at the calibration valve assembly.
7.4.2 The analyzer output shall be adjusted to the appropriate levels, if necessary.
7.4.3 The predicted response for the low-level and mid-level gases shall be calculated based on a linear response line between the zero and high-level responses.
7.4.4 Low-level and mid-level calibration gases shall be introduced successively to the measurement system.
7.4.5 The analyzer responses for low-level and mid-level calibration gases shall be recorded, and the differences between the measurement system responses and the predicted responses shall be determined. These differences must be less than ±5% of the respective calibration gas value. If not, the measurement system shall be deemed not acceptable and must be replaced or repaired prior to testing. No adjustments to the measurement system shall be conducted after the calibration and before the drift determination found in 8.2 of this appendix.
7.4.6 If adjustments are necessary before the completion of the test series, the drift checks shall be performed prior to the required adjustments, and the calibration following the adjustments shall be repeated.
7.4.7 If multiple electronic ranges are to be used, each additional range must be checked with a mid-level calibration gas to verify the multiplication factor.
7.5 Sampling System Bias Check
7.5.1 Perform the sampling system bias check by introducing calibration gases at the calibration valve installed at the outlet of the sampling probe. A zero gas and either the mid-range or high-range gas, whichever most closely approximates the effluent concentrations, shall be used for this check, as follows:
7.5.2 Introduce the upscale calibration gas and record the gas concentration displayed by the analyzer on a form approved by the Department. Then introduce zero gas, and record the gas concentration displayed by the analyzer. During the sampling system bias check, operate the system at the normal sampling rate, and make no adjustments to the measurement system other than those necessary to achieve proper calibration gas flow rates at the analyzer. Alternately introduce the zero and upscale gases until a stable response is achieved. The tester shall determine the measurement system response time by observing the times required to achieve a stable response for both the zero and upscale gases. Note the longer of the two times as the response time.
7.5.3 The sampling system bias check shall be considered invalid if the difference between the gas concentrations displayed by the measurement system for the analyzer calibration error check exceeds ±5% of the span for either the zero or upscale calibration gas. If an invalid calibration is exhibited, take corrective action, and repeat the sampling system bias check until acceptable performance is achieved. If adjustment to the analyzer is required, first repeat the analyzer calibration error check, then repeat the sampling system bias check.
7.6 Response time test.
7.6.1 Zero gas shall be introduced into the measurement system at the calibration valve assembly.
7.6.2 When the system output has stabilized, the owner or operator shall switch quickly to the high-level calibration gas.
7.6.3 The time shall be recorded from the concentration change to the measurement system response equivalent to 95% of the step change.
7.6.4 The test shall be repeated three times and the results averaged.
8.0 Emission measurement test procedure.
8.1 Organic measurement.
8.1.1 Sampling shall begin at the start of the test period.
8.1.2 Time and any required process information shall be recorded, as appropriate.
8.1.3 Periods of process interruption or cyclic operation shall be noted on the recording chart.
8.2 Drift determination.
8.2.1 Immediately following the completion of the test period and hourly during the test period, the zero and mid-level calibration gases shall be introduced, one at a time, to the measurement system at the calibration valve assembly. No adjustments to the measurement system shall be made until after both the zero and calibration drift checks are made.
8.2.2 The analyzer response shall be recorded.
8.2.3 If the drift values exceed the specified limits, the test results shall be invalidated preceding the check, and the test shall be repeated following corrections to the measurement system.
8.2.4 Alternatively, the test measurement system may be recalibrated as in 7.4 of this appendix and the results reported using both sets of calibration data (i.e., data determined prior to the test period and data determined following the test period).
9.0 Organic concentration calculations.
The average organic concentration shall be determined in terms of ppmv propane by the integration of the output recording over the period specified in the applicable provisions of this regulation.
10.0 Quality assurance.
10.1 The owner or operator shall assure proper calibration, maintenance, and operation of the continuous emissions monitoring system on a continual basis.
10.2 The owner or operator shall establish a quality assurance program to evaluate and monitor performance on a continual basis. The following checks shall be done routinely:
10.2.1 A daily calibration check for each monitor. The calibration shall be adjusted if the check indicates the instrument's calibration drift exceeds the specification established in 6.0 of this appendix.
10.2.2 A daily system audit that includes the following:
10.2.2.1 A review of the calibration check data.
10.2.2.2 An inspection of the recording system.
10.2.2.3 An inspection of the control panel warning lights.
10.2.2.4 An inspection of the sample transport/interface system (e.g., flowmeters, filters), as appropriate.
10.2.3 A quarterly calibration error test at the span midpoint.
10.2.4 The entire performance specification test repeated every second year.
11.0 Reporting of total hydrocarbon levels.
11.1 The total hydrocarbon concentration (THC) levels from the initial compliance certification test shall be reported as ppm propane for inlet and outlet concentrations and as a percent reduction across the control device.
11.2 THC levels shall be expressed in milligrams per second (mg/sec) (pounds per second [lb/sec]).
11.3 This conversion shall be accomplished using the following equation:
THC ppm propane = The total hydrocarbon concentration as actually measured by this method in ppm propane at the inlet or outlet;
Stack gas flow = Measured in dry standard cubic feet per second as determined by the flowmeter system or Methods 2 and 4;
5.2x10-2 = Constant to account for the conversion of units.
12.0 References.
12.1 Measurement of Volatile Organic Compounds--Guideline Series. U. S. Environmental Protection Agency, Research Triangle Park, North Carolina. Publication No. EPA-450/2-78-041. June 1978. p. 46-54.
12.2 Traceability Protocol for Establishing True Concentrations of Gases Used for Calibration and Audits of Continuous Source Emission Monitors (Protocol No. 1). U. S. Environmental Protection Agency, Environmental Monitoring and Support Laboratory. Research Triangle Park, North Carolina. June 1973.
12.3 Gasoline Vapor Emission Laboratory Evaluation--Part 2. U. S. Environmental Protection Agency. Office of Air Quality Planning and Standards. Research Triangle Park, North Carolina. EMB Report No. 75-GAS-6. August 1975.
12.4 Appendix D: Performance Specifications for Continuous Emissions Monitoring of Total Hydrocarbons in Hazardous Waste Incinerators, Boilers and Industrial Furnaces. Federal Register. 54:206 pp. 43743-43745. October 26, 1989.
1. Mention of trade names or specific products does not constitute endorsement by the Department or the U.S. EPA.
1.0 CEMS quality control (QC) program.
Each owner or operator of a CEMS shall develop and implement a CEMS QC program. At a minimum, each QC program shall include written procedures that describe in detail step-by-step procedures and operations for each of the following:
1.1 Initial and routine periodic calibration of the CEMS.
1.2 Calibration drift (CD) determination and adjustment of the CEMS.
1.3 Preventative maintenance of the CEMS (including spare parts inventory).
1.4 Data recording, calculations, and reporting.
1.5 Accuracy audit procedures including sampling and analysis methods.
1.6 Program of corrective action for malfunctioning CEMS.
2.0 Determining out-of-control condition for the CEMS.
2.1 If either the zero (or low-level) or high-level CD exceeds 10% for five consecutive daily periods, the CEMS is out-of-control.
2.2 If either the zero (or low-level) or high-level CD exceeds 20%, the CEMS is out-of-control.
2.3 If the CEMS fails a performance audit (PA) performed consistent with the requirements of 40 CFR, Part 60, Appendix F (July 1, 1992), the CEMS is out-of-control, and the owner or operator shall take necessary corrective action to eliminate the problem. Following the corrective action, the source owner or operator shall reconduct the appropriate failed portion of the audit and other applicable portions to determine whether the monitoring system is operating properly and within specifications. Monitoring data obtained during any out-of-control period may not be used for compliance determination or meet any data capture requirements; however, the data can be used for identifying periods when there has been a failure to meet quality assurance/quality control criteria.
3.0 Determining the out-of-control time period for the CEMS.
3.1 The beginning of the out-of-control period is either of the following:
3.1.1 The time corresponding to the completion of the fifth consecutive daily CD check with CD in excess of two times the allowable limit.
3.1.2 The time corresponding to completion of the daily CD check preceding the daily CD check that results in a CD in excess of four times the allowable limit.
3.2 The end of the out-of-control period is the time corresponding to the completion of the CD check following corrective action that results in the CD's at both the zero (or low-level) and high-level measurement points being within the corresponding allowable CD limit (i.e., either two times or four times the allowable limit in 40 CFR, Part 60, Appendix B, July 1, 1992).
3.3 If the CEMS failed a PA, the beginning of the out-of-control period is the time corresponding to the completion of the failed audit test. The end of the out-of-control period is the time corresponding to a successful retest of the PA sample.
4.0 Recordkeeping.
The owner or operator shall keep the QC procedure described in 1.0 of this appendix in a readily accessible location for at least five years and shall make the procedure available to the Department upon verbal or written request.
5.0 Reporting.
Upon verbal or written request, the owner or operator shall submit to the Department a copy of all information and records documenting out-of-control periods including beginning and end dates and descriptions of corrective actions taken.
1.0 Applicability.
This method determines the length of the rolling material balance period used in the liquid-liquid material balance test method to measure the overall performance of volatile organic compound (VOC) emission control systems employing carbon adsorbers for solvent recovery as the control device. The rolling balance period obtained from this method is source-specific, taking into account the particular configuration and operating parameters of the emission source (process) and its emission control system. Although the use of the rolling material balance approach may be applied to other types of solvent recovery control devices such as refrigeration, the method herein for determining the appropriate rolling balance period is limited to carbon adsorber-type solvent recovery control devices.
2.0 Summary of Method.
Physical properties and usage are determined for the solvent or solvents used in the process, and configuration and operating parameters are identified for the emission source and its emission control system. This information is used to calculate the concentration of VOC in the outlet air of the capture unit, amount of VOC adsorbed on the carbon, maximum VOC loading on the carbon, unmeasured solvent holding capacity of the solvent recovery system, and unmeasured solvent holding capacity of the process unit. These values are then used to calculate the rolling material balance period.
3.0 Definitions.
As used in this appendix, all terms not defined herein shall have the meaning given them in the November 15, 1990 Clean Air Act Amendments (CAAA), or in 2.0 of this regulation.
“Maximum carbon adsorber holding capacity for solvent, VA,” means the quantity of solvent (expressed as volume of liquid solvent) adsorbed on the carbon in all the adsorbent beds of the solvent recovery system that is in adsorption equilibrium with the capture unit outlet air stream under the process operating condition of maximum solvent usage.
“Rolling balance period (RBP)” or “rolling material balance period (RMBP)” means the number of consecutive operating 24-hour material balances, NMBP, used to determine the quantities of solvent usage and recovery for computation of the recovery efficiency for the emission control system, ER.
“Unmeasured process holding capacity for solvent, VP,” means the aggregate volume contained within the emission source (process) such as reservoirs, surge tanks, and transfer piping where the volumetric quantity of solvent-bearing liquid varies during process operation and where the quantity of solvent in this volume cannot be reasonably measured or determined as part of the daily solvent material balance around the process and emission control system. For the purpose of this method, the portion of the solvent that remains continuously filled with liquid under all process operating conditions is not considered part of the unmeasured process holding capacity for solvent.
“Unmeasured solvent recovery system holding capacity for solvent, VR,” means the sum of the maximum carbon adsorber holding capacity, VA, and the unmeasured solvent recovery system support equipment holding capacity, VS.
4.0 Procedure.
Follow the procedure below and complete the worksheets using configuration and operating data specific to the process unit or units and VOC emission control system for which the rolling balance period determination is being made.
4.1 Solvent Properties. The purpose of this step is to determine representative values for the molecular weight and liquid density of the solvent or solvents used in the process.
4.1.1 Single Component Solvent. If a single component solvent (not a mixture) is used exclusively for the process unit or units served by the emission control system, then record the chemical name of the solvent, its molecular weight, and liquid density in lines 1a, 2, and 3, respectively, of Worksheet A of this appendix and proceed to 4.2 of this appendix.
4.1.2 Multiple Solvent or Solvent Mixtures. If multiple solvents or solvent mixtures are used, complete Worksheet A of this appendix. List the chemical names of the individual organic compounds, including the major components of the solvent mixtures, in block 1 of Worksheet A of this appendix. The organic constituents listed should account for at least 90% of the total solvent. Use equation (I-1) of this appendix to calculate the weighted average molecular weight, ML, and liquid density, dL, of the solvents used in the process unit or units. Record these values on lines 2 and 3, respectively, of Worksheet A of this appendix.
Xi = Fraction of total solvent for VOC i.
|
Solvent Composition
|
(100•xi)
|
(Mi)
(lb/lbmole)
|
(di)
|
|||||
|
lb/lbmole
|
||||||||
|
lb/lbmole
|
||||||||
4.2 Emission Source and Control System Description. In this step, the site-specific system design and operation information needed to determine the appropriate rolling balance period is developed and compiled in Worksheet B of this appendix. Complete Worksheet B of this appendix by providing the indicated information specific to the emission source process unit or units and emission control system for which this rolling balance period determination is being made. Unless otherwise specified, the information requested refers to the aggregate of all the process units of the emission source linked to and served by the emission control system containing the carbon solvent recovery control device.
4.2.1 Number of Process Units. Record the total number of process units (e.g., dip tanks, spray booths) connected to the capture system and manifolded to the solvent recovery control system on line 1 of Worksheet B of this appendix.
4.2.2 Solvent Usage. The maximum daily solvent usage reported on line 2 of Worksheet B of this appendix is the maximum total solvent throughput of the process unit or units that would be used in a day under peak production. The average daily solvent usage requested in line 3 of Worksheet B of this appendix is the annual total solvent throughput of the process unit or units divided by the number of days in the year the process unit or units was in operation.
4.2.3 Unmeasured Process Holding Capacity for Solvent. Estimate (+10 gallons) the unmeasured solvent holding capacity of each process unit served by the capture and solvent recovery systems in this analysis. Compute the aggregate total unmeasured volume of these process units, and then compare this total to 10% of the value listed in Worksheet B, line 3 (0.1xQ'SP), of this appendix for the average daily solvent usage. Enter on line 4 the smaller of the values of the estimated aggregate total unmeasured process holding capacity and 10% of the average daily solvent usage reported on line 3 of Worksheet B of this appendix.
4.2.4 Capture System Air Flow Rate. Report on line 5 of Worksheet B of this appendix the total volumetric air flow rate (adjusted to the standard conditions of 70°F and 1.0 atm) output from the capture system to the solvent recovery system.
|
gala
|
||
|
scfmb
|
||
a Maximum value limited to 10% of the value in line 3, (Q'SP).
b Standard conditions: 70°F and 1.0 atm.
4.2.5 Maximum VOC Concentration in Capture System Outlet. Use equation (I-2) of this appendix to estimate the maximum concentration of VOC in the outlet air stream of the capture unit (inlet (2)(3) air stream to the carbon adsorber beds of the solvent recovery control unit). If actual VOC concentrations (measured on the same capture unit under the current operating conditions within the past two years using an EPA approved test method) are available and their average exceeds the concentration calculated in equation (I-2) of this appendix, then record the average of the measured concentrations on line 6 of Worksheet B of this appendix; otherwise record the concentration from equation (I-2) of this appendix on line 6 of Worksheet B of this appendix.
 (I-2)
(I-2)CO = Concentration of VOC in outlet air of capture unit, ppmv.
QSP = Maximum daily solvent throughput to the process units, gal/day (Worksheet B, line 2, of this appendix).
dL = Solvent liquid density, lb/gal (Worksheet A, line 3, of this appendix).
GCU = Capture unit outlet air flow rate, scfm (Worksheet B, line 5, of this appendix).
4.2.6 Quantity of Carbon in Solvent Recovery System. Report the number of carbon adsorber beds (both online and offline for regeneration) in the solvent recovery system on line 7 of Worksheet B of this appendix. Determine the total mass of carbon in the solvent recovery system by summing the masses in each of the carbon adsorber beds, and enter the total on line 8 of Worksheet B of this appendix.
4.2.7 Maximum Solvent Loading on the Carbon. To estimate the maximum solvent loading (i.e., solvent holding capacity) for the total carbon in the solvent recovery control unit, use equations (I-3) and (I-4) of this appendix. Record the value calculated for VA in equation (I-4) on line 9 of Worksheet B of this appendix.
X = ln(CO)
b0 = -3.047598
b1 = 0.275410
b2 = -0.003271
b3 = -0.000486
CO = Maximum VOC concentration in capture unit outlet air, ppmv (Worksheet B, line 6, of this appendix).
WA = Amount of VOC adsorbed on the carbon, lb VOC/lb carbon.
VA = Maximum solvent loading on the carbon in the solvent recovery unit, gal.
WA = Amount of VOC adsorbed on the carbon (equation (I-3) of this appendix).
WC = Total quantity of carbon in the solvent recovery unit, lb carbon (Worksheet B, line 3, of this appendix).
dL = Solvent liquid density, lb/gal (Worksheet A, line 3, of this appendix).
4.2.8 Unmeasured Solvent Capacity in Support Equipment. In addition to the solvent adsorbed on the carbon, the solvent recovery system may contain time-varying quantities of solvent in its support equipment such as condensate tanks, decanters, and surge/collection tanks, which are not measured as part of the daily material balance. Estimate that portion of the volume in the support equipment over which the unmeasured quantity of solvent it contains may vary between material balances. Do not include the capacity where the solvent inventory remains essentially constant over time. Compare this estimated value of the unmeasured solvent holding capacity in the support equipment with the value corresponding to 10% of the average daily solvent usage (Worksheet B, line 3, of this appendix), and record the smaller of these two values as VS on line 10 of Worksheet B of this appendix.
4.2.9 Unmeasured Solvent Capacity for Solvent Recovery System. The unmeasured solvent holding capacity of the solvent recovery system is the sum of the maximum solvent loading on the carbon and the unmeasured holding capacity of the support equipment. On Worksheet B of this appendix, sum the values on lines 9 and 10 and record this sum on line 11.
5.0 Data Analysis and Calculations.
Rolling Material Balance Period Determination. Use equation (I-5) of this appendix and the values of the indicated variables from Worksheet B of this appendix to compute the appropriate rolling balance period (number of consecutive 24-hour operating period material balances) for this specific emission source and emission control system.
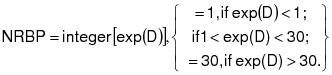 (I-5)
(I-5)b0 = 2.36877x100
QSP = Maximum daily solvent throughput to the process units, gal/day (Worksheet B, line 2, of this appendix).
VR = Unmeasured solvent holding capacity of the solvent recovery system, gal (Worksheet B, line 11, of this appendix).
VP = Unmeasured solvent holding capacity of the process units, gal (Worksheet B, line 4, of this appendix).
The methodologies presented in this attachment are based on the Ideal Gas Law and on fundamental vapor/liquid equilibrium relationships such as Henry's and Raoult's Law, unless otherwise specified. The equations are intended for use in estimating and characterizing uncontrolled emission streams from batch processes. Example calculations are presented for several unit operations. Significant errors may result in using the examples for situations that do not meet the conditions or assumptions that are clearly stated in the presentation of the methodologies.
1.0 Process Vent Emissions
Process vent emissions may result from a number of different events. Common process vents in batch processing result from drying, tank and reactor inert gas purging, vapor displacement losses from material transfer, tank and reactor heating, gas evolution, gas sparging, batch pressure filtration, and vacuum generation. The discussion below presents the principles and methodologies for estimating emissions during these events.
1.1 Drying
Two types of drying operations commonly occur in batch processing. These are conductive drying, in which heat is transmitted to the material to be dried by contact with heated surfaces, and convective drying, in which heat is transmitted by hot gases which are in contact with the material. Conductive drying may occur under vacuum conditions or at atmospheric pressure and in several types of dryers, including tray dryers, tumble (double-cone) dryers, pan dryers, and rotary dryers. Convective drying occurs at atmospheric pressure. Convective dryers include rotary dryers, fluid bed dryers, and spray dryers.
The methodology for calculating emissions from the types of dryers described above is essentially the same. In general, the rate of drying of the material depends on many factors associated with the specific drying situation (i.e., moisture or solvent content of material to be dried, heat and mass transfer parameters, drying period, etc.), but generally decreases with time so that a large percentage of the total liquid will be removed during the beginning of the drying cycle.
Studies on the theory of solids drying usually relate drying rate to moisture content of the solid. Three distinct periods of drying can be observed: the constant-rate period when surface moisture is evaporated; the funicular state when capillarity of the liquid in the pores influences the drying rate; and the pendular state when capillary action ceases and the liquid must diffuse through the pores of the solid. Each of these three drying periods has successively lower rates of drying; the final drying rate, when the moisture content is zero, is of course also zero (see 5.1 of this appendix).
Dryer designers often generate drying curves expressing the rate of drying as a function of residence time. Laboratory or pilot-scale experiments are often needed to establish the correct dryer size, operating temperature, gas flow rate, and cycle time. Once a dryer is installed and operating, readily available data include the cycle time, gas flow rates, and moisture content of the solid at the start and finish of the cycle. If very dry solids are produced (i.e., zero moisture content), the drying rate at the end of the cycle will be asymptotic to zero. This end point condition, and a knowledge of the total solvent removed, can be used to estimate the emission characteristics of an existing, installed dryer.
In industrial practice, the filter cakes and centrifuged solids to be dried often appear dry and free-flowing and may contain up to 50% solvent by weight. At the end of the drying cycle, the solvent content is reduced to a level required by the process. From a mass balance, the total amount of solvent emitted can be calculated.
To properly size emission control equipment for dryers, however, one must know the exhaust stream VOC content throughout the cycle. Although the precise VOC content can only be determined from extensive laboratory testing, a reasonable estimate can be obtained by assuming that the rate at which the drying rate decreases is linear over the length of the drying cycle starting from the initial, highest value and declining to zero at the end of the cycle. From the material balance on solvent removed, the average drying rate can be calculated knowing the length of the cycle. With a straight line relationship and a final value of zero, the initial drying rate must be two times the average. Therefore, the drying rate, and hence the emission rate, can be estimated for any point in the drying cycle.
1.1.1 Vacuum Dryers. Tray dryers, double-cone dryers, pan dryers, and rotary dryers may be operated under vacuum conditions. Such vacuum dryers will have an inward leakage rate of air that will aid in transporting the VOC's and air toxics that have evaporated from the dryer's product. Vacuums in the range of 15 to 25 inches mercury are typical. Articles have been published which provide methods of estimating leakage rates for vacuum systems (see 5.2 of this appendix). One such methodology is contained in 1.8 of this appendix. For a single dryer, the air leakage may range from 10 to 30 scfm depending on system design and vacuum level desired. An example calculation of vacuum dryer emissions follows:
Example: Consider the following example of a double-cone dryer operating at 15 inches of mercury, with an air-leakage rate of 15 scfm. The temperature inside the dryer is 60°F. Three hundred pounds of product cake, initially containing 25% by weight acetone are dried to less than 1% by weight solvent over the course of eight hours. Calculate the maximum VOC emission rate.

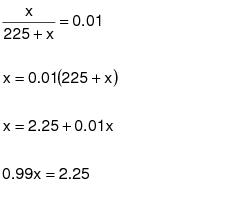
The airflow (leakage) is given as 15 scfm where 359 scf (at 0°C and 1 atm) is 1 mole. Therefore, the molar flow rate is:
1.1.2 Atmospheric Dryers. Convective dryers, such as tray and fluid bed dryers, operate at or above atmospheric pressure. The four types of conductive dryers discussed previously, tray dryers, double-cone dryers, pan dryers and rotary dryers may also be operated at atmospheric conditions. A stream of air or inert gas is used to move the volatilized material from the dryer vessel in conductive drying. The gas stream serves as the heating mechanism in convective drying. In both of these situations, the calculation of total VOC content emitted during the drying cycle is identical to the vacuum drying method (i.e., a mass balance from initial cake concentration to final cake concentration). The estimation of the maximum dryer emission rate, which is used for sizing of equipment, is analogous to the method presented for vacuum drying, with the flow rate of gas through the system equal to the dryer exhaust gas rate. An example calculation of atmospheric dryer emissions follows:
Example: A tray dryer uses 6,000 acfm of heated air (65°C) over a period of six hours to remove isopropyl alcohol (IPA) from a batch of solids. Each batch consists of 1,000 pounds of material containing 40% (by weight) solvent. The final product contains less than 0.6% solvent. Calculate the total uncontrolled VOC emissions per drying cycle and the maximum VOC emissions rate.
Solution: Mass balance over the drying cycle:
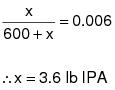
Knowledge of, or an estimate of (as above), the uncontrolled outlet stream composition is necessary to select an appropriate control technology. One should note that the mole fraction of the VOC is considerably lower (approximately two orders of magnitude) in the convective oven exhaust than in the vacuum oven (previous example).
1.2 Tank and Reactor Purging
In batch processing a gas stream is frequently used to purge VOC vapors from either an empty tank or reactor vessel, or from the vapor space of a partially filled tank or reactor. Typical reasons for purging are to maintain product quality (e.g., by using a dry sweep gas to minimize water vapor in a system) or to dilute flammable vapor concentrations below safety limits.
1.2.1 Empty Tank Purging. Empty vessels may be purged intermittently (e.g., at startup and shutdown, or between batches) using a displacement purge to remove accumulated vapors. The estimation of VOC emissions in this case is fairly straightforward. Before the purge, the initial VOC concentration of an empty vessel's vapor space is assumed to be equivalent to vapor in equilibrium with the removed liquid. The final concentration is a function of the number of purge gas volumes used. This relation can be expressed as a power law:
Cf = final concentration in vessel; and
Ci = initial concentration in vessel.
The fractional dilution per volume change assuming perfect mixing has been shown to be 37%. Thus, equation (K-1) of this appendix becomes:
This equation does not account for evaporation of any residual liquid in the vessel, and no free liquid. The equation was derived via the following steps:
Ca = concentration of VOC species
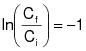 (K-5)
(K-5) Emissions are calculated by multiplying the vessel volume by the difference between final and initial concentrations, (Cf-Ci).
1.2.2 Filled Vessels. Filled or partially filled vessels are often "blanketed" with inert gas (or even air in the case of nonflammable solvents) using either "balanced pressure" or "trickle" control schemes (see 5.3 of this appendix). With balanced pressure blanketing, there is no flow of gas unless the tank liquid level changes (during filling or emptying) or the pressure rises or falls due to thermal effects. The calculation of emissions from this type of blanketing is analogous to "working and breathing" losses. For trickle blanketing, a constant flow of gas is maintained through headspace. The flow rate may be quite low for a storage tank, but may be much higher for a reactor where removal of water vapor or excess solvent vapor is required. The higher flows are referred to as purges or sweeps.
The volatile organic content of a purge gas stream may be calculated by assuming that the gas is saturated with the vapors of the liquid over which it is flowing. This assumption is generally conservative in that the VOC content of a gas cannot possibly be greater than saturation concentrations (as long as there are no entrained droplets). To calculate a maximum expected uncontrolled emissions rate, this approach is acceptable. However, the actual VOC concentration of the exiting purge gas may be substantially below saturation. Calculations show that the percent saturation of an inert gas purge stream over a quiescent pool of liquid is expected to be no more than 10%. The purging of equipment with a turbulent liquid surface leads to higher saturation fractions, approaching 100% saturation at lower flow rates. Since most vessels are typically agitated, the conservative assumption of complete VOC saturation is recommended at low to moderate purge gas flow rates. However, if the purge flow is greater than 100 scfm, a saturation factor of 25% is recommended.
Most operations are run at conditions, such as atmospheric pressure and relatively low temperatures, which allow the application of the ideal gas law. The VOC emission rate from purging may be estimated by first calculating the volumetric flow rate of the gas leaving the vessel, consisting of noncondensibles as well as the volatilized VOCs. The total rate of gas exiting a vessel is therefore:
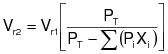 (K-6)
(K-6)Vr2 = rate of gas displaced from the vessel (VOC and noncondensibles), scfm;
Vr1 = rate of purge gas (noncondensibles), scfm;
PT = vessel pressure, mmHg;
ER = mass emission rate;
Yi = mole fraction in vapor phase, calculated in Equations (K-9) and (K-10) to follow in this appendix.
PT = pressure of the vessel vapor space; and
1.3 Vapor Displacement Losses--Transfer of Material to Vessels.
Emissions occur as a result of vapor displacement in many batch operations. The transfer of liquids from one vessel to another vessel causes a certain volume of gas to be displaced in the receiving vessel. The VOC's that may be contained in this volume also are displaced. In many cases, the displaced gas is vented directly to the atmosphere. The amount of VOC's emitted during such an event is limited by the partial pressure of the components in the gas stream and the vessel pressure. Usually, vessel vapor spaces are filled with air (i.e., 21% oxygen, 79% nitrogen) or an inert gas, such as nitrogen.
The degree of vent gas saturation with the VOCs must be assumed or known before any calculations are performed. When permit levels are established, a conservative assumption is typically made to prevent a low estimate of emissions. In most vapor displacement calculations one may conservatively consider the gas being displaced to be 100% saturated with the volatile compounds that are entering the vessel. The following steps are involved in calculating emissions from vapor displacement events:
The rate of displacement of a gas from a vapor space is equal to the rate of vessel filling with liquid. An example of this displacement is the transfer of liquid material from one process vessel to another, such as the charging of a reactor with material from a weigh tank, and the subsequent emission reactor gas that is saturated with the vaporized liquid.
The vapor pressure of the compound of interest (for one specific component, such as an air toxic) or of the entire volatile component of the liquid (for total VOC emissions) must first be determined. For one component, this can be done by consulting vapor pressure tables at the appropriate temperature or by using Antoine's equation, a form of which is presented below:
Pi* = vapor pressure of component i (mmHg);
There are several forms of vapor pressure estimation equations and the reader should be certain that the constants correspond to the appropriate form and that the units are consistent. Most physical property handbooks contain the Antoine equation and the appropriate constants.
If more than one compound is present in the liquid, the vapor pressure of all compounds in the mixture must be determined. After the vapor pressures have been determined, the partial pressure that the VOC vapor fraction exerts in the vessel vapor space may be determined by using Raoult's Law, which is a simple expression that describes equilibrium between an ideal vapor and an ideal liquid. The general equation for Raoult's Law is presented below:
Yi = mole fraction of i in the vapor
Pi = partial pressure of component i;
Xi =mole fraction of component i in the liquid;
Pi*=vapor pressure of component i at temperature T; and
PT =the total pressure in the vessel vapor space.
Raoult's Law may be used for multicomponent systems, assuming the compounds are completely miscible in one another. If the compounds are not miscible, or are only partially miscible, then they are considered "nonideal" and Raoult's Law does not apply. At or above the solubility limit, each compound exerts a partial pressure in the vapor space which is equal to the vapor pressure at that temperature. Below the solubility limit, especially for dilute solutions comprised of water and trace amounts of air toxics or VOC's, Henry's Law is used to describe the relationship between the mole fraction of the compound in the liquid and the vapor phase mole fraction. Henry's Law is presented below:
Xi=mole fraction of component i in the liquid;
Hi=Henry's Law constant for i (at temperature T);
Yi=mole fraction of component i in vapor; and
PT=the total pressure in the vessel vapor space.
This relationship is especially important in calculating evaporative losses from process wastewater.
Once Yi, the mole fraction of component i in the vessel vapor space, is known, the VOC or air toxic emission rate may be easily calculated by multiplying Yi by the vessel fill rate (which equals the gas displacement rate) and converting this volumetric rate to a mass emission using equation (K-7) of this appendix.
1.4 Vessel Heating
When a process vessel partially filled with a VOC or a material containing a VOC is heated, the gas and vapors in the headspace expand and are discharged from the vent. An estimate of the emissions in the uncontrolled vent stream from such an event can be calculated based on application of the Ideal Gas Law and on vapor-liquid equilibrium principles.
The basis of the calculation is that the moles of gas displaced from a vessel are a result of the expansion of the noncondensible gas upon heating, and an increase in the VOC vapor pressure. The assumptions made for the calculations which follow are (1) atmospheric pressure of 760 mmHg; and, (2) the displaced gas is always saturated with VOC vapor in equilibrium with the liquid mixture.
Pa1 = initial partial pressure of gas in vessel headspace, mmHg, and
(Pi)T1 = initial partial pressure of each VOC in vessel headspace, mmHg, at the initial temperature (T1).
The choice of formula to calculate Pi depends on which vapor-liquid equilibrium assumption is chosen (as discussed in 1.3 of this Appendix). If the VOC species behaves "ideally" in the system under consideration, then Raoult's Law holds and
Xi = mole fraction of each compound in the liquid mixture.
If the VOC in question is present in very dilute concentrations in the liquid, then Henry's Law gives a reasonable estimate of the compound partial pressure if the empirically determined constant is available:
Xi = Mole fraction of each compound in the liquid mixture.
Note: If neither Raoult's Law nor Henry's Law is considered to be valid for the compound mixture being considered, a more complex procedure, beyond the scope of this document, must be used. Commercial computer programs are available to simplify the task of calculating vapor-liquid equilibria for nonideal mixtures.
The calculation of Pi is repeated at the final temperature conditions, T2; and the final partial pressure of the gas in the vessel is calculated:
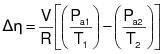 (K-15)
(K-15)R = Gas Law constant, 998.9 mmHg ft3/lb-moles °K;
Pa1 = initial gas pressure in the vessel, mmHg;
Pa2 = final gas pressure, mmHg;
The concentration of the VOC in the gas displaced at the beginning of the event is calculated assuming equilibrium at the initial vessel temperature. The final concentration of the VOC in the final amount of air displaced is calculated assuming equilibrium at the final vessel temperature. The VOC concentration in the displaced gas may be approximated by assuming it is equal to the average of the initial and final values. The number of moles of VOC displaced is equal to the moles of gas displaced times the average VOC mole fraction, as follows:
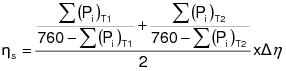 (K-16)
(K-16)çs = lb-moles of VOC vapor displaced from the vessel being heated up.
The mass of VOC vented can be calculated by multiplying the number of moles by the molecular weight. The reader should note that, at the boiling point of the VOC, this equation goes to infinity. In a physical sense, the vessel vapor space is filled entirely with VOC during boiling; the rate of release of VOC is therefore equal to the total flow of VOC out of the kettle. Therefore, this equation is not valid at the boiling point of the VOC.
1.5 Gas Evolution
When a gas is generated as the result of a chemical reaction, emissions may be calculated by assuming that the gas is saturated with any VOCs that are in contact with it at the exit temperature. Emission calculations are analogous to the filled vessel purging calculations and are calculated using the following formula to first calculate the rate of gas displaced:
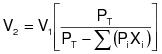 (K-17)
(K-17)V1 = initial volumetric rate of noncondensible gas evolution
PT = vessel pressure
(PiXi) = sum of the products of the vapor pressure and the mole fraction of each VOC at the exit temperature
Once V2 is known, it can be substituted into Equation (K-7) of this appendix to calculate the emission rate.
1.6 Sparging
Sparging is the subsurface introduction of a gas (typically nitrogen or other inert gas) intended to remove by selective volatilization (stripping) a more volatile minor component from a liquid mixture of predominantly less volatile material. Common applications of sparging are the removal of trace quantities of water or volatile organic solvent from a low volatility (high boiling point) resin. The removal of low concentrations of organic solvents from wastewater also may be achieved using air sparging.
Sparging is a semi-batch operation. The sparge tank is filled or discharged on a batch basis, while the gas is fed continuously at a steady flow rate for the duration of the sparge cycle. The subsurface sparger is designed to develop a mass of small diameter bubbles. The tank may be agitated as well in order to produce fine bubbles and increase the bubble residence time. These design features are intended to increase contact efficiency.
Utilizing fundamental chemical engineering principles and empirical correlations published in the literature it is possible to calculate the mass transfer coefficients encountered in sparging applications. The transfer rate of the component being stripped out is a function of temperature, composition, liquid diffusivity, gas rate and agitator power (which determine bubble size), and tank geometry (which, along with agitation power, determines residence time).
For the calculation of equilibrium concentration of VOC in the exiting sparge gas the earlier discussion of Raoult's Law and Henry's Law applies. For simple sparging (low viscosity fluids; no solids) vapor concentration may approach 100% of the calculated equilibrium value. For complex sparging, an empirically determined lower value may need to be used.
Unlike continuous flow vapor-liquid separation processes, with batch-wise sparging it is not possible to write a series of simple analytical equations which define the outlet gas concentration as a function of inlet concentration and thermodynamic properties of the compounds. This is because the liquid flow rate is zero and the composition changes with time. The problem of estimating the gas composition (hence, VOC emission rate) at any time during the sparge cycle, or of determining the amount of sparge gas and sparge item required to achieve a certain concentration reduction, can, however, be solved using simple numerical integration. One chooses a small time increment, one minute, for example, over which to calculate the gas flow and composition, making the simplifying assumption that the liquid composition does not change. From the inlet gas concentration (most likely zero) and the saturated exit gas concentration, the amount of volatile removed from the bulk liquid can be calculated, and a new estimate made for the liquid composition. The calculation of the vapor composition for the next item "slice" will be made based on this new liquid composition value. The cumulative quantity of volatile removed is used for subsequent estimates of the liquid composition.
A graphical representation of the vapor or liquid composition as a function of sparge time has a characteristic hyperbolic shape where the composition is asymptotic to zero. The initial composition is high, as is the stripping rate, because the mass transfer is a function of the composition driving force. The final composition is low, but a long stripping time is required to achieve a small decrease in composition because the driving force is also very low. An example sparging volatilization calculation follows:
Example: A 1,000-gal tank of wastewater containing a 0.025 wt% toluene is to be air sparged to remove the toluene to a concentration level of less than 20 ppb (by weight) to permit discharge to a municipal sewer system. Ambient air is to be used; the design temperature is 20°C. Toluene-water vapor-liquid equilibrium at 20°C can be approximated using a Henry's Law constant of 370 atm.
Solution: Use one minute time slices, assume a sparge rate, calculate time required to achieve concentration objective, adjust sparge rate until reasonable cycle time is calculated. Because of standard geometry of 1,000-gal tank, and modest gas rates, 100% of equilibrium concentration can be assumed. Table K-1 summarizes the results of the calculations made using a personal computer spreadsheet program. With 75 acfm of sparge gas, the desired concentration of 20 ppb toluene is achieved in 55 minutes of sparging. The table clearly shows that the bulk of the VOC is removed during the early part of the cycle: one-half of the total toluene is removed in the first three minutes, and 90% is removed after 13 minutes.
1.7 Batch Pressure Filtration
Pressure filtration of non-aqueous, volatile, flammable, or hazardous slurries is typically conducted in a closed vessel. Generally, VOCs are not emitted during the filtration step, as there is no venting from the process vessel. However, during the gas blowing (cake-drying) step of the cycle, or during pressure release prior to cake discharge, venting occurs and there is potential for VOC emissions.
The gas blowing step is intended to accomplish some preliminary cake drying by evaporating some of the liquid filtrate present in the filter cake. This operation is roughly equivalent to the constant drying-rate period of a dryer's operation except that heated gas is not used (except in the case of some special purpose equipment where heated gas is, in fact, used). The blowing gas follows the same flow path as the filtrate, so that it could be vented through the receiving tank.
1.7.1 Filter Cake Purging. The emission rate in the vented purge/blowing gas can be calculated if the cake conditions at the start and end of this portion of the cycle are known. The filtrate will be evaporated at approximately a constant rate. Assuming that the filtrate is 100% VOC, the emissions rate is simply:
Xi = the weight fraction of filtrate at the start of the gas-blowing step;
Xf = the weight fraction of filtrate at the end of the gas-blowing step;
However, one key piece of data required for the above calculations, namely the filtrate content of the cake before the gas blowing, is not usually available. Therefore, this methodology is of only limited utility.
Since the blowing gas causes the VOCs in the filtrate to evaporate, the gas stream is partially saturated with vapor, and approaches vapor-liquid equilibrium as a limit. An assumption of the attained percent saturation enables the calculation of an emission rate.
1.7.2 Depressurization. Prior to opening a batch pressure filter for solids discharges, the pressure must be relieved. In the case of a filter design utilizing a closed vessel, there is some compressed gas in the vapor space which will have some degree of vapor saturation of VOC from the filtrate. Upon depressurization, a fraction of the noncondensible gas along with the VOC vapor will be vented. The estimation of the emission rate from a depressurization event is a straightforward application of the Ideal Gas Law if certain simplifying assumptions are made. If the vessel has been under pressure for some time during the filter cycle, and no additional noncondensible gas has been added, then it is reasonable to assume that the gas is saturated with the VOC vapor at the vessel temperature. To simplify the calculations, one assumes that the pressure decreases linearly with time once depressurization has begun, and that the composition of the gas/vapor mixture is always saturated with VOC vapor through the end of the depressurization. The estimation of the emission rate proceeds according to the following steps:
1.7.2.1 Calculate the mole fraction of each VOC vapor species initially present in the vessel at the end of the depressurization.
Pi = vapor pressure of each VOC component i;
Xi = mole fraction of component i in liquid; and
Yi = mole fraction of component i initially in the vapor.
1.7.2.2 The moles of VOC initially in the vessel are then calculated using the Ideal Gas Law as follows:
P1 = initial vessel pressure;
1.7.2.3 The moles of noncondensible gas present initially in the vessel are calculated as follows:
1.7.2.4 At the beginning of the depressurization, there are more moles of noncondensible gas in the vessel relative to the moles of VOC in the vessel than at the end of depressurization. At the beginning of the depressurization, there are:  moles of VOC to noncondensables.
moles of VOC to noncondensables.
1.7.2.5 At the end of depressurization, there are: 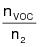 moles of VOC to noncondensables, where:
moles of VOC to noncondensables, where:
1.7.2.6 The moles of VOC for the duration of the depressurization may be calculated by taking an approximation of the average ratio of moles of VOC to moles of noncondensibles and multiplying by the total moles of noncondensibles released during the depressurization, or:
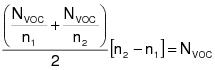 (K-23)
(K-23)NVOC = moles of VOC emitted.
1.7.2.7 The moles of VOC emitted can be converted to a mass rate using the following equation:
ERVOC = emission rate of the VOC;
MWVOC = molecular weight of the VOC; and
1.8 Emissions from Vacuum Generating Equipment
Steam ejectors and vacuum pumps are used to pull vacuums on vessels and can be sources of VOC and air toxic emissions. Both come in contact with a stream of gas that potentially contains pollutants. A steam ejector consists essentially of a steam nozzle that produces a high-velocity jet across a suction chamber connected to the vessel being evacuated. The gas from the vessel is entrained into the motive steam as it passes across the suction chamber. Both gas and steam are usually routed to a condenser.
Conventional (mechanical-type) vacuum pumps use a high boiling point oil to lubricate the moving parts. The VOCs which are present in the gas on the suction side may be partially condensed in the elevated pressure inside the vacuum pump. This reduces the amount of VOC emitted in the gas discharge from the pump, but causes contamination (reduced viscosity) of the pump oil. For this reason, if a significant amount of VOC is expected in the gas being evacuated, a liquid ring vacuum pump may be selected.
In a liquid ring vacuum pump, the vacuum is created by the rotating motion of a slug of seal fluid inside the pump casing. The seal fluid is in intimate contact with the gas and VOC being evacuated. A portion of the seal fluid is ejected with the pump discharge, so a system for seal fluid recycle and makeup is required.
Because the seal fluid is in contact with the gas/VOC mixture, mass and heat transfer can occur inside the pump. The emissions from a liquid ring vacuum pump are, therefore, a function of the seal fluid temperature and composition, as well as the inlet gas composition. For purposes of calculation one may assume that the exiting gas is in equilibrium with the seal fluid. The seal fluid must be chosen to be compatible with the gas/VOC being evacuated. In some cases, the seal fluid itself is a VOC and equilibrium with the exiting gas may result in an increase in VOC level from that in the suction side. In other cases, the seal fluid can act to reduce the VOC level of the gas stream by absorbing (or condensing, in the case of a cooled seal fluid system) some of the VOC in the gas being evacuated.
1.8.1 Emission Estimation. Emissions from vacuum systems originate from two distinct sources: 1) the first is the gas at the vacuum system discharge, 2) the second is the seal fluid or motive steam. Calculating emissions from the gaseous discharge of systems that serve to induce vacuums on equipment involves the estimation of the amount of air that leaks into the equipment because of the pressure differential between the inside and outside of the vessel. Once this air leakage rate is known, the rate of VOC emissions may be calculated by knowing the vacuum system discharge outlet temperature and pressure.
1.8.1.1 Air Leakage Estimation
The air leakage rate for the equipment may be estimated from the following equations, which correspond to the leakage created by metal porosity and cracks and leakage resulting from seals and components in a system for various vacuum pressure ranges:
1.8.1.1.1 Leakage from metal porosity and cracks
1.8.1.1.2 Leakage from seals and components
0 = specific leakage rate for components, lb/hr/in (presented in Table A-5 of Appendix A of EPA-453/R-93-017).
1.8.1.1.3 The total air leakage rate, La, in lb/hr, is merely the sum of the two components W and w.
Once the air leakage rate is known, the VOC emission rate, in lb/hr, may be calculated using the following equation from the 1978 Pharmaceuticals CTG (see 5.4 of this appendix):
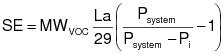 (K-32)
(K-32)MWVOC = molecular weight of VOC, lb/lb-mole;
Psystem = absolute pressure of receiving vessel or ejector outlet conditions, if there is no receiver;
Pi = vapor pressure of the VOC at the receiver temperature, in mmHg;
Calculating emissions from seal fluid or motive steam is analogous to the calculations of VOC emissions from other sources of wastewater, which is discussed below.
2.0 Evaporative Losses From Wastewater
Evaporative losses from wastewater that is contaminated with VOCs has been examined in detail, but currently is not included in this appendix. Several publications are available to aid the readers in calculating emissions from wastewater treatment systems which include surface impoundments, lagoons, and basins (see 5.5, 5.6, and 5.7 of this appendix).
3.0 Storage Tank Emissions
In general, emissions of VOCs from storage tank working and breathing losses appear to be no different for continuous processes than they are for batch processes. Both types of losses usually are calculated using EPA-derived storage tank loss equations for three types of storage tanks: fixed roof, external floating roof, and internal floating roof. Fixed roof and horizontal pressure tanks appear to be the most common storage vessels used in batch processing. Estimation equations for these tank types and a detailed explanation of their use, may be found in an EPA reference (see 5.8 of this appendix).
4.0 Equipment Leaks
The calculation of emissions of VOCs from leaking process line components such as valves, pump seals, flanges, and sampling connections is no different for continuous processes than it is for batch processes. Emissions tend to be lower because the amount of time that components are actually in VOC service is less for batch processes than for continuous processes. In the event that no other specific data is available, equipment leak emissions may be estimated using the equipment leak factors derived for the Synthetic Organic Chemical Manufacturing Industry (SOCMI). These factors are readily available, and are included in Table K-2 of this appendix. It is also possible to develop unit-specific emission estimates according to an accepted EPA protocol. The methodology for developing a specific emission estimate for leaking components is contained in another reference (see 5.10 of this appendix).
a These factors are appropriate for estimating emissions when no other data (i.e., leakage rates) are available. Source: EPA-953/R-93-026. June 1993 (see 5.9 of this appendix).
5.0 References
5.1 McCabe, W., and J. Smith. Unit Operations of Chemical Engineering, Third Edition. 1976.
5.2 Ryan, J.L., and S. Croll. Selecting Vacuum Systems. Chemical Engineering. 88:78. December 14, 1981.
5.3 Blakely, P. and G. Orlando. Using Inert Gasses for Purging, Blanketing, and Transfer. Chemical Engineering. 91:97-102. May 28, 1984.
5.4 EPA-450/2-78-029. Control of Volatile Organic Emissions from Manufacture of Synthesized Pharmaceutical Products. December 1978.
5.5 EPA-450/3-87-026. Hazardous Waste Treatment, Storage, and Disposal Facilities (TSDF) Air Emission Models.
5.6 Wastewater and Wastes Enabling Document. Version 1.0, July 1992.
5.7 Industrial Wastewater Volatile Organic Compound Emissions–Background Information for BACT/LAER Determinations.
5.8 AP-42 Compilation of Air Pollution Emission Factors, Chapter 12.
5.9 EPA-453/R-93-026, Protocol for Equipment Leak Emission Estimates. June 1993.
5.10 EPA-450/3-88-010, Protocol for Generating Unit-Specific Emission Estimates for Equipment Leaks of VOC and VHAP. October 1988.
1. Material in this appendix has been derived from guidance in Chapter 3 and Appendix C of EPA's draft CTG for Batch Processes (EPA-453/R-93-017).
1.0 Applicability and Principle.
1.1 Applicability. This method is applicable for the determination of organic carbon in diluted offset lithographic solutions.
1.2 Principle. Organic carbon in a sample is converted to carbon dioxide (CO2) by catalytic combustion or wet chemical oxidation. The CO2 formed can be measured directly by an infrared detector or converted to methane (CH4) and measured by a flame ionization detector. The amount of CO2 or CH4 is directly proportional to the concentration of carbonaceous material in the sample.
2.0 Sensitivity and Interferences.
2.1 Sensitivity. The method is most applicable to measurement of organic carbon above 1 mg/L.
2.2 Interferences. All distilled water used in making or diluting the samples must be acidified with concentrate H3PO4 (1 mL of H3PO4/1 L of water) and purged with inert gas (He, N2...) for at least 30 minutes. Inject this water into the Total Organic Carbon analyzer and determine the total concentration (ppm C) of the blank. This method is sufficient for removing most interferences due to inorganic carbon in the water. Do not purge the sample with an inert gas since purging may result in the loss of volatile organic substances.
3.0 Apparatus.
3.1 Blender. Waring-type or similar, for blending or homogenizing samples.
3.2 Total Organic Analyzer. An analyzer capable of measuring carbonaceous material in liquid samples. Consideration should be given to the types of samples to be analyzed, the expected concentration range, and the forms of carbon to be measured.
3.3 Volumetric Flasks and Volumetric Pipets. For preparing standard solutions and the lithographic solutions.
3.4 Glass Bottles. For sample collection and storage.
4.0 Reagents.
4.1 Water. Distilled water used in preparation of standards and for dilution of samples should be ultrapure to reduce the carbon concentration of the blank. Carbon dioxide-free, double-distilled water is recommended. Ion-exchanged waters are not recommended because of the possibilities of contamination with organic materials from the resins.
4.2 Potassium Hydrogen Phthalate, Stock Solution. 1000 mg carbon/L. Dissolve 0.2128 g of potassium hydrogen phthalate (Primary Standard Grade) in distilled water and dilute to 100.0 mL.
4.3 Potassium Hydrogen Phthalate, Standard Solutions. Prepare standard solutions from the stock solution by dilution with distilled water.
4.4 Blank Solution. Use the same distilled water (or similar quality water) used for the preparation of the standard solutions.
5.0 Sample Preparation.
5.1 Prepare the offset lithographic solutions according to the manufacturer's directions.
5.2 Dilute the offset lithographic solutions with H2O to be within the calibrated range of the instrument before analyzing. Dilutions of 1-to-100 or greater may be necessary before lithographic solutions can be analyzed.
6.0 Procedure.
6.1 Follow the instrument manufacturer's instructions for calibration, procedure, and calculations.
6.2 Calibrate using at least three standards. The set of calibration standards should consist of one below the expected concentration, one above the expected concentration, and one approximately at the expected concentration.
6.3 Calculate and report the results as mg C/g sample.
7.0 Precision and Accuracy.
7.1 Precision and accuracy determinations for diluted offset lithographic solutions were determined by the method of standard addition. Two analysts in one laboratory analyzed spiked and unspiked diluted solutions. The results are contained in 8.4 of this appendix.
7.2 Precision and accuracy determinations for diluted lithographic cleaning solutions were determined by the method of standard addition. One analyst in one laboratory analyzed spiked and unspiked diluted solutions. The results are contained in 8.4 of this appendix.
8.0 Bibliography.
8.1 Annual Book of ASTM Standards, Part 31, "Water", Standard D 2574-79, p. 469 (1976).
8.2 Standard Methods for the Examination of Water and Wastewater, 14th Edition, p. 532, Method 505, (1975).
8.3 Method 415.1, Methods for Chemical Analysis of Water and Wastes, Environmental Monitoring and Support Laboratory, USEPA, Cincinnati, OH 45268, EPA 600/4-79-020.
8.4 Evaluation of Method 415.1 for Offset Lithographic Solutions. September, 1992.
This appendix presents a test method for evaluating the performance of alternative cleaning fluids. Any fluids may be tested, but the primary intent is that it will be used to evaluate the performance of alternatives relative to a VOC solvent. It is a screening technique designed to determine whether the alternative (or alternatives) cleans at least as well as a currently used VOC solvent in a simple, standardized wiping application. The results of this procedure may not mimic those that would be achieved for a different scenario in an industrial setting (e.g., spraying or wiping a complex shape). However, any cleaning fluids that are unsatisfactory in this test can be eliminated from consideration for more complicated site-specific tests.
1.0 Standard Test Method For Determining the Performance of Alternative Cleaning Fluids.
Introduction. Industrial plants use VOC solvents to clean numerous contaminants from a variety of materials in different configurations. Alternative solvents and cleaning fluids exist that would produce lower VOC emissions from many of these cleaning applications. This method involves comparative testing of an existing VOC solvent with alternatives using one standardized cleaning procedure. It is a screening technique that identifies which alternative fluids clean as well as or better than an existing VOC solvent. Because it may not reproduce the plants' actual cleaning procedure, nor determine the effect of the alternative on the performance of coatings applied to the cleaned surface, it is likely that additional site- or industry-specific tests will be needed before the alternatives that pass this screening test are adopted.
The method is based on ASTM Method D 4828-91 for determining the practical washability of organic coatings. Changes were made to the method to allow its use in new applications. The changes include a wider variety of acceptable test panel materials, contaminants, and cleaning fluids. Procedures for evaluating the results are also different. The cleaning apparatus and procedure were not modified.
2.0 Applicability and Principle.
2.1 Applicability. This method applies to the determination of the relative ease of removal of contaminants from a variety of materials/surfaces by manual or mechanical cleaning with a sponge and various solvents or other cleaning solutions.
2.2 Principles. A contaminant is applied to a test panel to represent a typical industrial cleaning situation. One portion of the soiled panel is scrubbed with a sponge and the existing solvent, and another portion is scrubbed with a sponge and an alternative solvent or cleaning solution that produces lower VOC. The performance of the alternative is then rated as (1) worse than the existing solvent, or (2) as good as or better than the existing solvent.
3.0 Apparatus.
3.1 Sponge and Holder;
3.2 Contaminant Applicator;
3.3 Weight, 100 g;
3.4 Balance, Weighing Accurately to 0.1 g;
3.5 Doctor or Bird Film Applicator, having a 7-mil (0.018-mm) clearance by 6-in. (150-mm) film width;
3.6 Panels of various materials, 17½ by 6½ by ¼ in. (455 by 165 by 6.3 mm);
3.7 Washability Machine3;
3.8 Masking Tape;
3.9 Straightedge, approximately 17 in (430 mm) in length;
3.10 Cotton Tipped Swabs;
3.11 Medicine Droppers;
3.12 Suction Plate, for drawdowns.
4.0 Reagents and Materials.
4.1 Contaminants. Examples that may be used with this test method include, but are not limited to pencil, crayon, ball-point pen, waterborne felt-tip markers, grease, and mineral oil.
4.2 Solvents and Cleaning Solutions. Examples that may be used with this test method include any VOC solvent or alternative cleaning fluid.
4.3 Test Panels. Different types of panels may be selected to match the cleaning application. Examples include, but are not limited to, glass, stainless steel, aluminum, and plastic. The surface may be painted or unpainted.
5.0 Preparation of Apparatus.
5.1 Washability Machine. Level the apparatus before use and operate at 37 ± 1 cpm. (A cycle consists of a complete forward and reverse stroke.)
5.2 Sponge and Holder. Add sufficient weight to the holder in the form of metal sheets or other flat weights to give a combined weight of 1,000 g, including the dry sponge.
5.3 Test Panel. Prepare paint coated panels by the following procedure. Stir the material thoroughly and strain, if necessary, to remove all skins and particles. Draw down the coating on the panel. Apply the coating in 3 to 4 s from end to end to prevent pin holes or holidays in the film. Air dry all panels in a horizontal position for seven days in a room maintained at 73 ± 3.5°F (23 ± 2°C) and 50 ± 5% relative humidity as described in Specification D3924, or under conditions specifically applicable to the material under test. Prepare enough panels with each paint for all the projected tests. Before use, clean the top of the test panel (painted or unpainted) to be sure it is free of specks.
6.0 Procedure.
6.1 Application of Contaminants.
6.1.1 Apply the selected contaminants to the test panel (or coating on the panel) in one straight line parallel to the length of the panel for the manual method of cleaning, or in a pair of lines perpendicular to the length of the panel for the mechanical method of cleaning.
6.1.2 Apply solid contaminants using the apparatus shown in Figure M-1 of this appendix. Insert pencil, crayon, pen or similar items into the appropriately sized hole and secure its position so it extends 1½ in. (40 mm) beyond the panel (see Figure M-1(a) of this appendix). Secure the medium in position with a piece of masking tape (see Figure M-1(b) of this appendix). Put the wooden applicator panel at one end of the test panel and place the 100-g weight on its top face at the end nearest to the marking device, as shown in Figure M-1(b) of this appendix, securing it with a piece of tape. Allow the nonweighted end of the wooden applicator panel to rest on the surface of the test panel, then hold it by the outer edges and pull it along the entire length of the panel (see Figure M-1(c) of this appendix).
6.1.3 Apply liquid contaminants using hand-held cotton-tipped swabs. Immerse one end of a cotton-tipped swab in an appropriate solvent or cleaning solution and allow to remain totally immersed until the cotton tip is saturated (approximately 10 to 15 sec). Remove the tip from the liquid and apply the first of two parallel lines to the test panel using the straightedge to assist in drawing the lines. Adjustment of pressure on the cotton tip may be required to provide a line of uniform intensity. Reimmerse the cotton tip in the liquid and then draw the second line. Repeat with a clean or unused cotton tip for each liquid being used. Permit the contaminants to dry at least one hour under the same temperature and humidity conditions as in 5.3 of this appendix.
Note 1 - Only one contaminant may be tested at one time. Typically, as noted above, this will mean the application of one line for manual cleaning or two parallel lines of contaminant for mechanical cleaning. As shown in Figure M-2 of this appendix, one section of the panel will be used to test an alternative cleaning fluid. However, the panel may be long enough to allow evaluation of more than one alternative cleaning fluid in a single test.
6.2 Cleaning.
6.2.1 Soak the sponge in the solvent or solution at ambient temperature until saturated. Remove the sponge and squeeze with one hand until no more liquid drips from the sponge. Replace the sponge in the holder and pour 15±1 mL of solvent or cleaning solution on the exposed face of the sponge.
6.2.2 Apply five mL of solvent or cleaning solution in parallel bands to each contaminant line.
6.3 Manual Method. Place the sponge and holder atone end of the panel so that its long axis is perpendicular to the length of the panel (see Figure M-2 of this appendix). Rub the sponge across the panel over the contaminant lines, exerting minimum downward pressure. Continue rubbing until all the contaminants are removed or to a maximum of 100 cycles. If all the contaminants are removed prior to 100 cycles, stop and record the number of cycles before proceeding to 6.5 of this appendix.
6.4 Mechanical Method.
6.4.1 Place the sponge and holder at one end of the panel so that its long axis is parallel to the length of the panel (see Figure M-2 of this appendix). Attach the sponge and holder to the cable of the washability machine. Allow the sponge to travel a maximum of 100 cycles. If all the contaminants are removed prior to 100 cycles, stop and record the number of cycles before proceeding to 6.5 of this appendix.
6.4.2 Remove the test panel and evaluate the condition of each in the path of the sponge and rate as follows:
6.4.2.1 Worse than existing solvent.
6.4.2.2 As good as or better than existing solvent. When a contaminant is removed prior to 100 cycles, note the number of cycles in which each contaminant was removed.
7.0 Report. Report the Following Information:
7.1 Type of contaminants, solvents, or cleaning solutions, and washing method used and the results obtained in 6.5 of this appendix.
7.2 Any contaminants that were removed in less than 100 cycles.
7.3 Any deviation from the recommended procedure.
8.0 Precision and Bias
8.1 Precision - Unknown.
8.2 Repeatability - Unknown.
8.3 Reproducibility - Unknown.
8.4 Bias - Unknown.
1 Material in this Appendix has been derived from Appendix F of Alternative Control Techniques Document—Industrial Cleaning Solvents, EPA-453/R-94-015.
2 A sponge, 3 by 3¼ by 1¾ in. (75 by 95 by 45 mm), Part No. AG-8116, and a metal holder, Part No. AG-8115, available from BYK-Gardner, Inc., 2435 Linden Lane, Silver Spring, MD 20910 or a sponge, Part No. WA 2222, and metal holder, Part No. WA 2220, available from Paul N. Gardner Co., 316 N.E. First Street, Pompano Beach, Florida 33060-6699 have been found acceptable for this purpose. An equivalent may be used.
3 Washability machine, Model AG-8100, available from BYK-Gardner, Inc. or Model WA 2037D, available from the Paul N. Gardner Co., have been found suitable for this purpose. Other straight-line wash testers may be adapted to meet the requirements of this test method emissions.